
Benutzer:OnkelDagobert:Archiv:Innere Medizin:Herz
Grundlagen
[Bearbeiten]
- Anatomie des Herzens
-
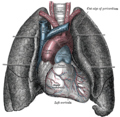 Lage des Herzens in situ
Lage des Herzens in situ -

-
 Querschnitt durchs Herz
Querschnitt durchs Herz -

-
Darstellung der linken Koronararterie mittels Herzkatheter




- Physiologie des Herzens
-
 Blutfluss durchs Herz
Blutfluss durchs Herz -
 Erregungsausbreitung im Herzen und Beziehungen zum EKG
Erregungsausbreitung im Herzen und Beziehungen zum EKG -
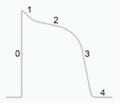 Potenzialverlauf eines Myozyten
Potenzialverlauf eines Myozyten -
 Potenzialverlauf von Zellen des Sinusknotens
Potenzialverlauf von Zellen des Sinusknotens -
Druckverlauf im linken Ventrikel während eines Herzzyklus
-
 Druck-Volumen-Diagramm eines Herzzyklus
Druck-Volumen-Diagramm eines Herzzyklus




{kind=link}
{kind=link}
Bei der Aortenisthmusstenose behindern kongenitale Membranen der Aortenwand, die sich meist gegenüber dem Ductus arteriosus Botalli befinden (d. h. an der Aortenhinterwand), die Blutströmung. Gefäßabschnitte vor der Stenose weisen einen erhöhten Blutdruck auf (mit Nachlaststeigerung und konsekutiver Linksherzhypertrophie), Gefäßabschnitte nach der Stenose einen verringerten Blutdruck mit entsprechenden klinischen Auswirkungen. Man unterscheidet eine infantile und eine adulte Form:
- Bei der infantilen Form befinden sich die Gefäßmembranen auf Höhe (juxtaduktal) oder proximal der Ductus-Einmündung (präduktal). Oft tritt die Störung zusammen mit anderen kardiovaskulären Defekten auf.
- Bei der adulten Form liegen die Gefäßmembranen distal der Ductus-Einmündung (postduktal).
Bei der Aortenstenose wird der Blutfluss im Bereich der Aortenklappe (subvalvulär, valvulär, supravalvulär) behindert. Bildlich gesprochen muss das Herz erhöhte Kraft aufbringen, um das erforderliche Blutvolumen gegen den Widerstand zu fördern; als Reaktion hierauf hypertrophiert das Myokard (konzentrische, später auch exzentrische Linksherzhypertrophie mit Behinderung der Kammerfüllung). Dennoch ist die geförderte Blutmenge nicht ausreichend, was zu Perfusionsstörungen der Koronarien und der Organe führt. Letztlich kommt es hierdurch zum Bild einer Linksherzinsuffizienz (Schwindel, Synkopen) mit Angina pectoris.
Bei der koronaren Herzkrankheit (ischämische Herzerkrankung) sind die Koronargefäße nicht mehr in der Lage, den Sauerstoffbedarf des Herzmuskels zu decken, da entweder das über die Koronargefäße bereitgestellte Sauerstoffangebot verringert ist (aufgrund von [intra- oder extraluminal bedingten] Verengungen der Koronargefäße, Verringerung des Perfusionsdrucks oder Hypoxämie) oder aber der Sauerstoffbedarf des Herzens erhöht ist (wegen erhöhter Herzarbeit [Herzmuskelhypertrophie, Tachykardien, Hypertonie] oder erhöhter Wandspannung [chronische Druck- oder Volumenbelastungen]). Die Sauerstoff-Unterversorgung des Herzens löst einen Sauerstoffmangelschmerz aus (Angina pectoris; typischerweise ausgelöst durch Belastung, sistiert in Ruhe oder durch Applikation von Nitroglyzerin [stabile Angina pectoris]). Die Zellen stellen ihren Stoffwechsel auf anaerobe Glykolyse um, die aufgrund der zunehmenden Azidose jedoch letztlich gehemmt wird, so dass es zur Nekrose (Koagulationsnekrose) mit konsekutiver Narbenbildung kommt (Infarkt). Durch die Elektrolytverschiebungen, die infolge der Ischämie auftreten, können maligne Herzrhythmusstörungen entstehen.
Das akute Koronarsyndrom umfasst drei Entitäten:
- Myokardinfarkt (STEMI, NSTEMI),
- instabile Angina pectoris,
- plötzlicher Herztod.
Die instabile Angina pectoris wird deshalb unter dem Krankheitsbild subsummiert, da ihre Symptome kaum von denen des Myokardinfarkts zu unterscheiden sind. In erster Linie geht es hierbei um den Myokardinfarkt. Dieser entsteht meist auf dem Boden einer koronaren Herzerkrankung. Seltener spielen andere Erkrankungen wie Vaskultiden, Aortendissektion, kongenitale Koronaranomalien eine kausale Rolle. Meist handelt es sich um Vorderwandinfarkte, seltener um Hinterwand- oder Seitenwandinfarkte. Je nach dem, ob alle Wandschichten oder nur das innere Drittel des Myokards betroffen sind, spricht man von einem transmuralen oder einen Innenschichtinfarkt. Die Symptomatik ist dramatisch: retrosternaler, persistierender (und nitrorefraktärer) Vernichtungsschmerz (ggf. mit Ausstrahlung) mit sympathikotoner Reaktion. Sind große Teile des Herzmuskels betroffen, kommt es zur akuten Herzinsuffizienz mit entsprechenden Symptomen. Bei Störung der vegetativen Innervation des Nervens (autonome Neuropathie, etwa bei Diabetes mellitus) kann sich ein Myokardinfarkt ohne Schmerzen präsentieren.
Damit es zu einem Infarkt kommt, muss die Sauerstoffversorgung des Myokards stark eingeschränkt sein, etwa bei starker Anstrengung oder erhöhter Thrombogenität des Blutes. Zunächst tritt eine Ischämie auf, in deren Folge es zu Elektrolytstörungen der Myozyten kommt und das Ruhemembranpotential weniger negativ wird; dies birgt das Risiko für Herzrhythmusstörungen. Durch ATP-Mangel und Insuffizienz der Natrium-Kalium-ATPase kommt es daraufhin zu Ödem, Ruptur und Nekrose der Myozyten. In der Folge wird eine Entzündungsreaktion (Einwanderung von Neutrophilen, später von Makrophagen) initiiert und Narbengewebe gebildet (zunächst Einsprossen von Granulationsgewebe, später Bildung der definitiven Narbe). Die Narbe kann sich später ausdehnen, was zur Bildung von Koronaraneurysmen und kardialer Funktionsverschlechterung führen kann.
Ein Myokardinfarkt kann zu schweren Komplikationen führen, akut vor allem zu Herzrhythmusstörungen. Bei ausgedehntem Infarktareal kommt es zur akuten Herzinsuffizienz. Bei Nekrose von Papillarmuskeln tritt eine akute Mitralinsuffizienz auf, bei Nekrose der Herzwand kann es zur Ruptur mit Bildung eines Hämoperikards und konsekutiver Perikardtamponade kommen. Im späteren Verlauf können unter anderem folgende Störungen auftreten: Dressler-Syndrom (Autoimmunperikarditis), Herzwandaneurysmen (die eine strukturelle Schwachstelle und Emboliequelle darstellen), chronische Mitralklappeninsuffizienz.
Bei den Endokarditiden unterscheidet man infektiöse von nicht infektiösen.
Infektiöse Endokarditiden:
Bei der akuten infektiösen Endokarditis wird das durch mechanische oder immunologische Faktoren (z. B. bei rheumatischen Erkrankungen) vorgeschädigte Endokard infiziert von Bakterien, Pilzen oder anderen Mikroorganismen, die in die Blutbahn eingedrungen sind. Folge: Entzündungsreaktion mit Degeneration der Klappen (polypöse Auflagerungen, Vegetationen), im weiteren Verlauf auch Störung der Herzfunktion. Einzelne Erreger oder größere Erregergruppen können sich ablösen, was zu Bakteriämie (mit protrahierter Infektsymptomatik) bzw. septischen Embolien mit Ausbildung von Organabszessen führt; immunologische Reaktionen können die Bildung von Immunkomplexen anstoßen, die zu Entzündungen in Gelenken, Gefäßen oder Nieren führen (Arthritis, Vaskulitis, Glomerulonephritis mit entsprechenden Folgen); durch die Vaskulitiden bilden sich auch die "Stigmata" der Endokarditis: Osler-Knötchen (Akren), Splinter-Hämorrhagien (subungual), Janeway-Läsionen (palmar oder plantar), Roth-Flecken (Retina).
Milder verläuft die subakute infektiöse Endokarditis (Endocarditis lenta), die meist durch niedrig virulente Streptokokken (Streptococcus viridans) ausgelöst wird, allerdings ebenfalls zu einer Degeneration der betroffenen Strukturen führt. Typisch ist der protrahierte, „schleichende“ (lenta) Verlauf (> 40 Tage).
Nicht infektiöse Endokarditiden:
Neben den infektiösen Endokarditiden gibt es eine Reihe von nicht infektiösen Endokarditiden, die im Rahmen von (meist immunologisch bedingten) Erkrankungen oder aufgrund angeborener Defekte entstehen:
- Endocarditis verrucosa rheumatica: Als Folge des rheumatischen Fiebers.
- Endocarditis thrombotica: Bei auszehrenden Erkrankungen.
- Endocarditis parietalis fibroplastica Löffler (eosinophile Endokarditis): Allergisch bedingte Endomyokarditis mit Fibrosierung; Infiltration des parietalen Endokards mit eosinophilen Granulozyten, zudem Bluteosinophilie. Auftreten im Rahmen des Löffler-Syndroms, bei dem auch weitere Organe betroffen sind.
- Endocarditis Libman-Sacks: Bei systemischem Lupus erythematodes.
- tropische Endomyokardfibrose: Genetisch bedingte Verdickung des Endokards.
- Degenerative Endokarditis: Sich von der Klappenbasis her ausbreitende kalzifizierende Klappendestruktion.
Kardiomyopathien (Erkrankungen des Herzmuskels) beruhen auf intrinsischen Defekten des Myokards (genetische Anomalien; primäre Kardiomyopathien) oder treten als Folge von kardialen oder extrakardialen Störungen auf (Infektionen, Immunprozesse, Ischämien etc.; sekundäre Kardiomyopathien). Unter diesem Begriff lassen sich eine Reihe von Störungen des Herzmuskels subsummieren. Im engeren Sinne unterscheidet man jedoch vier Formen (häufige Symptomatik: Herzinsuffizienz, Angina pectoris, Rhythmusstörungen):
- Dilatative (kongestive) Kardiomyopathie: Durch angeborene Defekte, jedoch auch aufgrund von Ischämien (KHK), Noxen (Alkohol) oder Entzündungen (Coxsackie-B-Virus) hypertrophiert das Myokard exzentrisch (Dilatation), wodurch sich die systolische Kontraktionsfähigkeit der Ventrikel verringert, so dass letztlich mehr Volumen im Ventrikel verbleibt und weniger ausgeworfen wird. Das Herz fördert somit weniger Blut, so dass es schließlich zu einer progredienten Herzinsuffizienz kommt. Die Dilatation der Myozyten prädisponiert zudem zu Rhythmusstörungen.
- Hypertrophe Kardiomyopathie: Aufgrund von genetischen Störungen hypertrophiert das Myokard (asymmetrische Hypertrophie; obstruktiv oder nicht obstruktiv), wodurch sich das Ventrikellumen einengt und somit auch das diastolische Volumen abnimmt; dies führt zu einer (diastolischen) Herzinsuffizienz. Ist zudem der Ausflusstrakt aufgrund der Hypertrophie verengt (obstruktive Form vs. nicht obstruktive Form), so ist eine funktionale Aortenstenose die Folge. Es können Rhythmusstörungen auftreten.
- Restriktive (obliterative) Kardiomyopathie: Meist aufgrund einer Endomyokardfibrose (tropische Endokardfibrose, Endocarditis parietalis fibroplastica Löffler) kommt es zu einer Myokardversteifung, wodurch sich die (diastolische) Dehnbarkeit der Ventrikel verringert; z. T. ist auch das Ventrikellumen eingeengt. Die Folge: Herzinsuffizienz.
- Arrhythmogene rechtsventrikuläre Kardiomyopathie: Genetisch oder viral bedingt tritt hier eine Degeneration des Myokards auf, die neben hämodynamischen Störungen zu einer elektrischen Instabilität der Myozyten führt, auf deren Boden maligne Herzrhythmusstörungen entstehen können.
Rhythmusstörungen
[Bearbeiten]

Schema:
- Beurteilung von Rhythmus und evtl. Arrhythmien
- Bestimmung der Herzfrequenz
- Bestimmung des Lagetyps
- Bestimmung der Zeiten (PQ-Intervall, QRS-Intervall, QT-Intervall)
- Beurteilung von Zacken und Strecken: P-Welle, QRS-Komplex, ST-Strecke, T-Welle
Beurteilungskriterien (Auswahl):
- Rhythmus: Sinusrhythmus (Arrhythmia absoluta bei Vorhofflimmern)?
- Lagetyp: Indifferenz- oder Steiltyp (überdrehter Linkstyp bei links-anteriorem Hemiblock, Rechtstyp bei links-posteriorem Hemiblock, Rechtsherzbelastung)?
- P-Welle: 1:1-Assoziation mit QRS-Komplex (nicht bei Vorhofflattern, AV-Blocks ab II°)? Negativ in I oder II (AV-Knoten-Ersatzrhythmus)? Verlängert (AV-Block I°) oder verkürzt (WPW-Syndrom)? Form (sägezahnartig bei Vorhofflattern)?
- Q-Zacke: Amplitude erhöht (Zeichen eines abgelaufenen Infarkts)?
- R-Zacke: R-Progression von V1 bis V4 (fehlend bei abgelaufenem Vorderwandinfarkt)?
- QRS-Komplex: Hinweise auf Schenkelblock (kompletter Schenkelblock: QRS in V1 [RSB] oder V6 [LSB] verbreitert, M-Form, T-Negativierung)?
- ST-Strecke und T-Welle: ST-Hebung (akuter Infarkt, Perikarditis)? ST-Senkung, T-Negativierung (Myokardischämie, Linksherzhypertrophie, Digitalis etc.)?
Man unterscheidet bradykarde (verringerte Herzfrequenz) und tachykarde (erhöhte Herzfrequenz oder zusätzliche Erregungen) Herzrhythmusstörungen, wobei man die tachykarden Rhythmusstörungen in supraventrikuläre (Vorhöfe einschließlich AV-Knoten) und ventrikuläre Tachykardien einteilt. Ventrikuläre Tachykardien können tödlich ausgehen, supraventrikuläre Tachykardien und Bradykardien hingegen verlaufen in der Regel nicht letal (Ausnahme: Reentry-Tachykardie beim WPW-Syndrom).
Klinisch äußern sich tachykarde und bradykarde Rhythmusstörungen mit Zeichen der Herzförderinsuffizienz: Schwindel, Synkopen, Schwächegefühl bis hin zum plötzlichen Herztod. Tachykardien werden zudem häufig als Herzrasen oder Herzstolpern wahrgenommen.
Den Tachykardien liegen Störungen in Vorhof, Kammer oder Erregungsleitungssystem zugrunde. Bradykardien beruhen hingegen meist auf intrinsischen oder extrinsischen (nervalen, metabolischen, endokrinen) Störungen des Erregungsbildungs- und -leitungssystems.
Supraventrikuläre Tachykardien:
- Supraventrikuläre Extrasystolen: Bei Schädigung von Vorhofmyozyten auftretende Extrasystolen. Im EKG: vorzeitig einfallende, aber regelrecht konfigurierte Kammerkomplexe
- Sinustachykardie: Vor allem metabolische, endokrine oder toxische Faktoren führen zu einer (allmählichen) Steigerung des Sympathikotonus, der die Depolarisationsfrequenz des Sinusknotens erhöht.
- Vorhofflattern, Vorhofflimmern: Schädigungen von Vorhofmyozyten (v. a. Dehnung) sowie endokrine, metabolische oder andere Einflüsse führen über Spontandepolarisationen zur Entwicklung von einem (Vorhofflattern) oder mehreren kleinen (Vorhofflimmern) Reentry-Kreisläufen (sägezahnartige bzw. unruhige Nullinie im EKG). Beim Vorhofflattern (Frequenz ca. 300/min) wird die Erregung tachykard und regelmäßig auf die Kammern übergeleitet, beim Vorhofflimmern unregelmäßig (zum Teil tachykard oder bradykard: Tachy- bzw. Bradyarrhythmia absoluta). Bei länger dauerndern Vorhofflimmern kommt es zu strukturellen Veränderungen, die eine Chronifizierung begünstigen. Vorhofflimmern birgt zudem das Risiko von Schlaganfällen (Thrombenbildung im Vorhof aufgrund verlangsamten Blutflusses).
- AV-Knoten-Reentrytachykardie und WPW-Syndrom: In beiden Fällen liegen akzessorische Leitungsbahnen vor (Längsdissoziation des AV-Knotens bzw. Kentbündel). Das Einfallen von supraventrikulären bzw. ventrikulären Extrasystolen führt zu einem Reentry der Erregung von der physiologischen auf die zusätzliche Leitungsbahn und vice versa. Die Folge: Plötzlich einsetzende und ebenso plötzlich endende Tachykardie. Typisches EKG eines WPW-Syndroms: verkürzte PQ-Dauer, Deltawelle; im Anfall: deformiertes P nach dem QRS-Komplex. Sonderform: LGL-Syndrom (Jamesbündel).
Ventrikuläre Tachykardien:
- Ventrikuläre Extrasystolen: Vorzeitige Depolarisation von (geschädigten) Kammermyozyten. Im EKG: vorzeitig einfallende, verbreiterte QRS-Komplexe, kompensatorische postextrasystolische Pause.
- Anhaltende ventrikuläre Tachykardie: Veränderungen der Erregbarkeit von Myozyten sowie verlängerte Erregungszeiten (z. B. bei Narben nach Herzinfarkt) führen hier zu einem Reentry-Kreislauf im Kammermyokard.
- Torsade-de-Pointes-Tachykardie: (Angeborene) Störungen von Ionenkanälen sowie bestimmte Medikamente führen zu einer Verlängerung der QT-Zeit (Long-QT-Syndrom) mit verzögerter Repolarisation. Gewisse Konstellationen (Bradykardien, Hypokaliämie) haben frühe Nachdepolarisationen zur Folge, die die Torsade-de-pointes-Tachykardie auslösen. Im EKG: Tachykardie mit um die isoelektrische Linie drehender QRS-Achse ("Torsade de pointes").
- Kammerflattern, Kammerflimmern: Wie beim Vorhofflattern und -flimmern führen hier Myozytenschädigungen oder metabolische, endokrine oder andere Einflüsse zu einer elektrischen Instabilität mit konsekutiven Reentry-Kreisläufen.
Bradykardien:
- AV-Block: Strukturelle oder metabolisch verursachte Störungen des AV-Knotens führen zu einer verlangsamten (Grad I: PQ > 200 ms), teilweise (Grad II: Wenckebach [= Mobitz I]: zunehmende PQ-Verlängerung bis zum Ausfall einer Überleitung; Mobitz [= Mobitz II]: Ausfall der Überleitung in fixem Verhältnis) oder vollständig (Grad III mit Ersatzrhythmus nach Adam-Stokes-Anfall) ausgefallenen Erregungsüberleitung vom Vorhof- aufs Kammermyokard.
- Sick-Sinus-Syndrom, SA-Block: Störungen der Zellen des Sinusknotens oder der Überleitung von diesen Zellen auf den Vorhof (Grad-Einteilung wie beim AV-Block).
- Karotissinussyndrom: Überempfindlichkeit vagaler Afferenzen führen hier zu einer abnormen Verringerung des Sympathikotonus bei Stimulation der Barorezeptoren im Karotissinus. Man unterscheidet einen kardioinhibitorischen (Verringerung der Herzfrequenz) und deinen vasodepressorischen Typ (Druckabfall wegen abnormer Vasodilatation).
Blutdruck
[Bearbeiten]Der Blutdruck ist theoretisch das Produkt aus Widerstand und Herzminutenvolumen. Entsprechend unterscheidet man bei der arteriellen Hypertonie einen Widerstandshochdruck und einen Volumenhochdruck. Der Widerstandshochdruck kann beispielsweise durch einen erhöhten peripheren arteriellen Gefäßtonus oder durch erhöhte Blutviskosität hervorgerufen werden. Der Volumenhochdruck beruht entweder auf einer Steigerung der Herzfrequenz oder auf einer Erhöhung des Schlagvolumens, etwa infolge einer Erhöhung des Intravasalvolumens oder aufgrund einer Tonisierung der venösen Kapazitätsgefäße. In den meisten Fällen liegt eine primäre Hypertonie vor, die im Rahmen eines metabolischen Syndroms oder bei Nierenschädigung (verminderte Nephronzahl mit folglich vermehrter Salz- und Wasserretention) auftreten kann. Selten ist die Hypertonie sekundär, d. h. durch eine andere Krankheit bedingt. Es handelt sich dabei vorwiegend um Erkrankungen/Störungen des Nierenparenchyms (mit Salz- und Wasserretention) oder der renalen Perfusion (Nierenarterienstenose), um endokrine Erkrankungen (Phäochromozytom, Cushing-Syndrom, primärer Hyperaldosteronismus, Adrenogenitales Syndrom) oder kardiovaskuläre Störungen. Eine Hypertonie kann zudem medikamentöse oder Ernährungsursachen haben (z. B. Lakritzabusus). Die arterielle Hypertonie ist anfangs meist symptomarm, führt aber zu schwerwiegenden Komplikationen an Herz (Linksherzhypertrophie, KHK), Nieren (Nephrosklerose), Hirn (Schlaganfälle, Demenz), Gefäßen (Durchblutungsstörungen, Aortendissektion). Bei der malignen Hypertonie kommt es zu Enzephalopathie, Nephropathie und Retinopathie.