
Praktikum Anorganische Chemie/ Druckversion

Dieser Text ist sowohl unter der „Creative Commons Attribution/Share-Alike“-Lizenz 3.0 als auch GFDL lizenziert.
Eine deutschsprachige Beschreibung für Autoren und Weiternutzer findet man in den Nutzungsbedingungen der Wikimedia Foundation.
Sicherheitsvorschriften
[Bearbeiten]Bei allen Arbeiten im Labor muss immer eine Schutzbrille und ein Schutzkittel sowie geeignete Kleidung (lange Hose und geschlossene Schuhe) getragen werden. Gegebenenfalls muss zusätzlich eine Gummi- oder Lederschürze getragen werden. Gearbeitet wird in der Regel im Abzug. Zum einen verhindert man so das Austreten von Gasen und Dämpfen in die Raumluft und ist zusätzlich noch vor Spritzern usw. sicher. Bei Arbeiten, die bekanntermaßen sicher sind, wie z.B. Titrationen oder bei vielen Reaktionen in der Tüpfelplatte, kann auch auf dem Labortisch gearbeitet werden. Die Schutzscheibe des Abzugs muss so weit wie möglich geschlossen gehalten werden, damit er eine wirkungsvolle Sogwirkung entfalten kann und einen Spritz- und Explosionsschutz bietet. Ein Bunsenbrenner sollte in einem gewissen Abstand zur Scheibe aufgestellt werden, da eine dauerhafte thermische Belastung zu Rissen führen kann und wieder die Sicherheit gefährdet. Substanzen, die ätzende oder giftige Gase absondern, sollten auch unter dem Abzug nur in verschlossenen Gefäßen aufbewahrt werden.
Beim Umgang mit einigen Substanzen muss außerdem ein Gesichtsschutz verwendet werden (z. B. Flusssäure). Handschuhe sind beim Umgang mit giftigen oder ätzenden Stoffen angebracht. Sie sollten jedoch nicht dazu verleiten unsauber zu arbeiten. Außerdem ist nicht jedes Handschuhmaterial für jeden Stoff geeignet.[1] Vor allem Einmalhandschuhe sind als Schutz gegen Lösungsmittel gänzlich ungeeignet. Gegen wässrige Lösungen und manche Feststoffe bieten sie einen begrenzten Schutz. Handschuhe aus Nitril sind gegen manche Lösungsmittel, vor allem Aceton, unbeständig. Daher informiere man sich vor Arbeitsbeginn anhand der Beständigkeitsliste des Herstellers über die Eignung des Materials. Über die genauen Schutzmaßnahmen geben die Sicherheitsdatenblätter des Herstellers oder Händlers von Chemikalien Auskunft. Außerdem sind Betriebsanweisungen zu beachten!
- Gebotszeichen nach DIN 4844-2
-
 Augenschutz benutzen
Augenschutz benutzen -
 Handschutz benutzen
Handschutz benutzen -
 Schutzkleidung benutzen
Schutzkleidung benutzen -
 Gebrauchsanweisung beachten
Gebrauchsanweisung beachten




Falls besonders giftige Substanzen entstehen, wird in diesem Arbeitsbuch mit dem Totenkopf darauf aufmerksam gemacht. Das entbindet jedoch nicht vor der Pflicht, sich vor Beginn eines Versuches genau über die Gefahren-, Sicherheits- und Entsorgungshinweise zu informieren. Gefahrstoffe sind stets mit den Gefahrensymbolen und R- und S-Sätzen (Risiko und Sicherheit) zu kennzeichnen. Zuverlässige Internetquellen findet man unter Wikipedia:Redaktion Chemie/Quellen.
- Gefahrensymbole nach Richtlinie 67/548/EWG
-
 explosiv (E)
explosiv (E) -
 brandfördernd (O)
brandfördernd (O) -
 leichtentzündlich (F)
leichtentzündlich (F) -
 hochentzündlich (F+)
hochentzündlich (F+) -
 giftig (T)
giftig (T) -
 hochgiftig (T+)
hochgiftig (T+) -
 gesundheitsschädlich (Xn)
gesundheitsschädlich (Xn) -
 reizend (Xi)
reizend (Xi) -
 ätzend (C)
ätzend (C) -
 umweltgefährlich (N)
umweltgefährlich (N)










- UN/GHS-Piktogramm
-

-

-

-

-

-

-

-

-










Vor jeder Arbeitspause, nach dem Arbeitsende und natürlich auch bei direktem Kontakt mit Chemikalien gründlich die Hände waschen!

Alle Blei- und Quecksilbersalze sowie sechswertige Chromverbindungen sind giftig und sollten daher nur in kleinstmöglichen Mengen verwendet werden. Jeglicher Hautkontakt mit der Probe- bzw. Salzlösung ist zu vermeiden (evtl. Handschuhe), da vor allem Chromate sowie Nickel auch Kontaktallergien auslösen können. Sechswertige Chromverbindungen (Chromate und Dichromate) sind außerdem krebserregend (Kategorie I)! Entsorgung von Chromaten und Dichromaten: Im gekennzeichneten Becherglas sammeln und mit naszierendem Wasserstoff (aus der Reaktion von Salzsäure mit unedlem Metall, z.B. Zink) oder mit Ethanol in Gegenwart von Schwefelsäure über längere Zeit behandeln. Das hierbei entstehende grüne Chrom(III) entsprechend den anderen Schwermetallen entsorgen.
- ↑ Bericht der Berufsgenossenschaft Chemie über einen tödlichen Unfall bei Verwendung ungeeigneter Handschuhe
Qualitative Analyse
[Bearbeiten]Dies ist eine Liste von Nachweisreaktionen geordnet nach Reagenzien. Sie ist möglicherweise nützlich, wenn eine Nachweisreaktion nicht so klappt wie gewünscht und man nun auf der Suche nach einem Hinweis ist, was stattdessen reagiert hat.
| Eisen(III) | Cobalt | Quecksilber | |
|---|---|---|---|
| Thiocyanat | [Fe(SCN)(H2O)5]2+ stierblutrot |
Co2[H2O]5[SCN]] pink, nach Extraktion mit Pentanol blau |
Co[Hg(SCN)4]
kornblumenblauer Niederschlag |
| Kupfer(II) | Kupfer(I) | |
|---|---|---|
| Thiocyanat | Cu(SCN)2 schwarz |
Cu(SCN) weiß |
In ammoniakalischen Lösungen bilden sich folgende Färbungen/Niederschläge mit dem Chelatkomplexbildner Diacetyldioxim:
| Nickel | himbeerot |
|---|---|
| Kupfer | braunrot |
| Eisen(II) | rot |
| Cobalt | braunrot |
| Bismut | zitronengelb |
| Blei | weiß |
| Palladium | gelblich |
Blutlaugensalze
[Bearbeiten]| Eisen(II) | Eisen(III) | |
|---|---|---|
| K4[Fe(CN)6] gelbes Blutlaugensalz |
K[FeIIIFeII(CN)6]↓ Berliner Blau | |
| K3[Fe(CN)6] rotes Blutlaugensalz |
K[FeIIIFeII(CN)6] Turnbulls Blau |
| Kupfer | Zink | |
|---|---|---|
| K4[Fe(CN)6] gelbes Blutlaugensalz |
Cu2[Fe(CN)6]↓ roter Niederschlag |
K2Zn3[Fe(CN)6]2↓ weißer Niederschlag |
| K3[Fe(CN)6] rotes Blutlaugensalz |
Zn3[Fe(CN)6]2↓ gelb-brauner Niederschlag |
Iodidlösung
[Bearbeiten]| Blei | Bismut | Antimon | Quecksilber | |
|---|---|---|---|---|
| Iodidlösung | PbI2↓ gelber Niederschlag |
BiI3↓ schwarzer Niederschlag |
SbI3
gelb-oranger Niederschlag |
HgI2
roter Niederschlag |
| Iodidlösung Überschuss |
[PbI4]2– farbloser Komplex |
[BiI4]– orangefarbener Komplex |
[HgI4]2-
farbloser Komplex |
Vorproben
[Bearbeiten]Die Flammenfärbung ist eine Vorprobe für viele Kationen und besonders bei Alkali- und Erdalkalimetallen zum Nachweis geeignet. Die Flammenfärbung wird mit einem Bunsen- oder Teclubrenner durchgeführt. Die Brennertypen unterscheiden sich in der Form und der Bedienung leicht. Während beim Bunsenbrenner die unterschiedlichen Flammtypen durch Verstellen der Abdeckung über den Lüftungsschlitzen eingestellt werden, geschieht dies beim Teclubrenner durch Verstellen der Abdeckung an der Unterseite des Brennerkamins. Die wichtigsten Flammentypen sind die leuchtende Flamme, bei welcher die Luftöffnungen verschlossen sind und eine unvollständige Verbrennung stattfindet. Die Flamme ist durch verglühende Rußteilchen gelb gefärbt. Bei nahezu vollständig geöffneter Belüftung entsteht eine Flamme, die leicht blau gefärbt ist und in die Reduktionszone im inneren Kegel und die Oxidationszone im äußeren Kegel eingeteilt werden kann. Sie ist wesentlich heißer und ist die in der Regel verwendete Flamme bei beiden Brennertypen.
-
 Flammentypen in Abhängigkeit von der Luftmenge, die das Luftventil passiert:
Flammentypen in Abhängigkeit von der Luftmenge, die das Luftventil passiert:
1: Ventil geschlossen (Diffusionsflamme)
2: Ventil zur Hälfte geöffnet
3: Ventil fast vollständig offen
4: Ventil voll geöffnet (Vormischflamme) -
 Teclubrenner
Teclubrenner -
 Natrium-D-Linie durch ein Spektroskop bei 589 nm
Natrium-D-Linie durch ein Spektroskop bei 589 nm


Man nimmt einen Magnesiastab und glüht diesen 5 Minuten lang im Bunsenbrenner aus, bis die gelbe Farbe verschwindet. Es handelt sich um eine Natriumflammenfärbung, da alles, was man berührt mit kleinen Mengen Handschweiß kontaminiert ist. Danach nimmt man mit dem heißen Magnesiastab etwas Analysensubstanz auf und hält ihn in die Brennerflamme, am besten in einem abgedunkeltem Abzug. Durch die Farbe der Flamme kann man einen ersten Hinweis erhalten. Eine exakte Unterscheidung ist jedoch nur mit einem Handspektroskop möglich. Falls Natrium in der Probe ist, werden alle anderen Flammenfärbungen überdeckt, hier hilft ein Blick durch Cobaltglas, welches das intensive Natriumgelb herausfiltert.
Farben
[Bearbeiten]


Spektren
[Bearbeiten]
Erklärung
[Bearbeiten]
Durch die Wärmeenergie werden die Elektronen auf ein höheres Energieniveau befördert ( angeregter Zustand). Diese Energie geben sie aber oft schnell wieder ab und fallen auf ihr vorheriges Niveau zurück. Die Energie, die sie abgeben, wird in Form von Licht spezifischer Wellenlänge abgegeben und ist bei gleichen Niveauänderungen immer gleich. Deshalb kann dies zur Identifikation eines Elementes dienen. Mit einem Spektroskop, das das Licht in seine Spektralfarben (vgl. Regenbogen) aufbricht, kann man die charakteristischen Linienspektren erkennen. Was vom menschlichen Auge als eine Farbe wahrgenommen wird, ist in Wirklichkeit ein diskontinuierliches Spektrum mit einzelnen Banden.
Die Schmelzperle aus Borax oder Phosphorsalz ist eine beliebte Vorprobe für Kationen. Man sollte sich jedoch nicht gänzlich auf das Ergebnis verlassen, sondern es eher als Hinweis sehen und versuchen die Indizien durch spezifische Nachweisreaktionen zu erhärten.
Durchführung
[Bearbeiten]
Man erhitzt ein Magnesiastäbchen oder ein Öse vom Platindraht im Bunsenbrenner. Der Platindraht bringt in dieser Probe aber keinen Vorteil gegenüber dem Magnesiastäbchen. Nun tunkt man sie in ein wenig NaNH4HPO4 Natriumammoniumhydrogenphosphat oder Na2[B4O5(OH)4]·8 H2O Borax (Natriumtetraborat) und schmilzt diese in der Bunsenbrennerflamme, nimmt wieder ein wenig Salz und schmilzt weiter, bis man zu einer möglichst gleichmäßigen, durchsichtigen Perle gelangt. Diese stippt man direkt in die Analyselösung oder kurz in eine wenig verdünnte Salzsäure und dann in die Analysensubstanz. Dann wieder kurz in die Bunsenbrennerflamme halten und versuchen die Substanz in die Perle einzuschmelzen. Je nachdem ob man in der Oxidations- oder der Reduktionsflamme glüht, erhält man andere Färbungen. Die tiefblaue Cobaltperle ist am eindeutigsten, überdeckt jedoch auch alle anderen Perlenfärbungen.
Farben
[Bearbeiten]| Oxidationsflamme | ||||
|---|---|---|---|---|
| Phosphorsalz | Borax | |||
| heiß | kalt | heiß | kalt | |
| Chrom | dunkelgelb | grün | grün | grün |
| Mangan | violett | violett | violett | violett |
| Eisen | gelbrot | gelb | gelbrot | gelbrot |
| Cobalt | blau | blau | blau | blau |
| Nickel | rotbraun | gelb | rotbraun | farblos |
| Kupfer | grün | blaugrün | grün | blaugrün |
| Reduktionsflamme | ||||
|---|---|---|---|---|
| Phosphorsalz | Borax | |||
| heiß | kalt | heiß | kalt | |
| Chrom | grün | grün | grün | grün |
| Mangan | farblos | farblos | farblos | farblos |
| Eisen | grünlich | grünlich | orange | grün |
| Cobalt | blau | blau | blau | blau |
| Nickel | farblos | farblos | farblos | farblos |
| Kupfer | farblos | lackrot | grünlich | lackrot |
Erklärung
[Bearbeiten]Boraxperle
[Bearbeiten]Das Borax-Anion zerfällt beim Schmelzvorgang in Bortrioxid und Natriummetaborat:

Das Bortrioxid verbindet sich dann mit einem Metallkation zu einer farbigen Verbindung:
- Bortrioxid reagiert mit Cobaltsulfat zu Natriummetaborat, Cobaltmetaborat und Schwefeltrioxid

Phosphorsalzperle
[Bearbeiten]Beim Erhitzen kondensiert NaNH4HPO4 zu Polyphosphaten, z.B. in ringförmige Metaphosphate Na3(P3O9). In der Reaktionsgleichung wird vereinfachend von dem Monomer NaPO3 ausgegangen.
- Natriumammoniumhydrogenphosphat reagiert zu Natriummetaphosphat, Ammoniak und Wasser

Bei der Reaktion mit Sulfaten ergibt sich folgende Reaktionsgleichung
- Natriummetaphosphat und Cobaltsulfat reagieren zu Natriumcobaltphosphat und Schwefeltrioxid

Sicherheitshinweis
[Bearbeiten]Borax kann Schäden am ungeborenen Kind hervorrufen.

gelb: Chromat
Die Oxidationsschmelze eignet sich als Nachweis für Chrom und Mangan und wird auch als Aufschluss für säureschwerlösliche Stoffe verwendet.
Durchführung
[Bearbeiten]Die Substanz wird sehr fein gepulvert, mit der dreifachen Menge einer 1:1-Mischung von Soda ( Natriumcarbonat) und Kalisalpeter ( Kaliumnitrat) oder Chilesalpeter ( Natriumnitrat) vollständig vermischt und im Porzellantiegel oder auf der Magnesiarinne vorsichtig zur Schmelze gebracht.
Erklärung
[Bearbeiten]Oxidationsschmelze von Chrom-(III)-Oxid (grün) zu gelbem Chromat

Oxidationsschmelze von Eisen-(II)-Chromit zu gelbem Chromat und Eisen-III-oxid

Die Oxidationsschmelze eignet sich als Nachweis bzw. Vorprobe für Mangan. Sie kann auch als Aufschluss in größerem Maßstab im Nickeltiegel durchgeführt werden.
Durchführung
[Bearbeiten]Die Substanz wird mit einem Kaliumhydroxid-Plätzchen auf einer Magnesiarinne vorsichtig geschmolzen. Nach beendeter Reaktion und Vorhandensein von Mangan bildet sich ein, teils nur sehr leichter, grüner Rand auf der Rinne. Zur besseren Überprüfung wird die Schmelze mit Wasser in eine Porzellanschale gespült. Dabei bildet sich eine grüne Lösung. Versetzt man die Lösung vom Rand her mit konzentrierter Essigsäure, so färbt sie sich rosa und man beobachtet einen schwarzen Niederschlag. Am besten führt man hier eine Blindprobe durch.
Erklärung
[Bearbeiten]Mangan wird in der Schmelze bis zur Oxidationszahl +VI oxidiert. Das entstandene grüne Kaliummanganat ist in alkalischer Lösung stabil und disproportioniert in saurer Lösung in Permanganat und Mangan(IV) (Mangandioxid, Braunstein).
Aufschlüsse
[Bearbeiten]Metallkationen stören viele Anionennachweise und müssen daher vorher abgetrennt werden.
Durchführung
[Bearbeiten]Hierfür kocht man 0,1g der Ursubstanz mit der vierfachen Menge Soda und eineinhalb Reagenzgläsern Wasser etwa 15 Minuten lang. Dann lässt man die Lösung wieder abkühlen, damit möglichst vollständig ausgefällt wird. Man filtriert oder zentrifugiert die Lösung und verwirft den Rückstand. Durch leichtes Ansäuern und Erwärmen wird das enthaltene CO2 ausgetrieben. Aus dem klaren Filtrat können nun Anionennachweise durchgeführt werden.
Erklärung
[Bearbeiten]Es fallen die Kationen aus, die schwerlösliche Carbonate bilden:
- Metallsalz und Natriumcarbonat reagiert zu Metallcarbonat, Anion geht in Lösung

und diejenigen, die im alkalischen Milieu schwerlösliche Hydroxide bilden:
- Metallsalz und Natriumcarbonat reagiert zu Metallhydroxid und Hydrogencarbonat, Anion geht in Lösung

Der Soda-Pottasche-Aufschluss erlaubt die Überführung von Erdalkalisulfaten, hochgeglühten Oxiden, Silikaten und Silberhalogeniden in leichter lösliche Verbindungen. Durch die Verwendung eines Salzgemisches aus Soda und Pottasche wird der Schmelzpunkt gegenüber den reinen Verbindungen abgesenkt.
Das Tiegelmaterial wird durch die Zusammensetzung der Ursubstanz bestimmt.
Durchführung
[Bearbeiten]Zunächst versucht man, die Ursubstanz in Salzsäure zu lösen. Der unlösliche Rückstand wird mit destilliertem Wasser gewaschen und im Trockenschrank getrocknet. Die Substanz wird sehr fein gepulvert, mit der vier- bis sechsfachen Menge einer 1:1-Mischung von Soda ( Natriumcarbonat) und Pottasche ( Kaliumcarbonat) vollständig vermischt und im Porzellantiegel (nicht für Aluminiumoxid und Silikate) oder in einem Nickeltiegel (für Sulfate und hochgeglühte Oxide) vorsichtig bis zur klaren Schmelze erhitzt. Es ist zu beachten, dass dabei immer Verunreinigungen durch Aluminium und Silicium bzw. Nickel in die Analysensubstanz übergehen.
Nach dem Abkühlen wird die Schmelze gemörsert und in Wasser aufgenommen. Solange mit verdünnter Natriumcarbonatlösung waschen, bis das Filtrat sulfatfrei ist (keine Trübung von Bariumchloridlösung mehr).
Erklärung
[Bearbeiten]Erdalkalisulfate werden in Carbonate überführt:

Schwerlösliche Silikate werden in lösliches Natriumsilikat überführt:

Mit dem Freiberger Aufschluss ist ein Aufschluss für Zinn(IV)-oxid (SnO2, Zinnstein). Zinn bildet ein leichtlösliches Thiostanat. Er kann aber auch zum Aufschluss anderer Metalloxide, die leichtlösliche Thioverbindungen bilden, verwendet werden.
Durchführung
[Bearbeiten]Die Substanz, normalerweise der in Salzsäure unlösliche Rückstand, wird im bedeckten Porzellantiegel mit der sechsfachen Menge eines Gemisches aus Schwefel und wasserfreiem Natriumcarbonat (1:1) geschmolzen. Während der Reaktion entstehen Schwefeldioxid und Kohlendioxid. Hört die Gasentwicklung auf, ist die Reaktion beendet. Der Schmelzkuchen wird in verdünnter Natronlauge gelöst und unlösliche Bestandteile abgetrennt. Die Lösung wird dann mit verdünnter Salzsäure angesäuert. Dabei fallen die entsprechenden Sulfidverbindungen aus. Sie können dann mit den entsprechenden Nachweisen nachgewiesen werden.
Erklärung
[Bearbeiten]Aufschluss von Zinndioxid

Der saure Aufschluss wird verwendet um basische oder amphotere Metalloxide, vor allem Eisen(III)-oxid Fe2O3, Chrom(III)-oxid Cr2O3 und Aluminiumoxid Al2O3, aufzuschließen.
Durchführung
[Bearbeiten]Die Substanz wird mit der sechsfachen Menge Kaliumhydrogensulfat verrieben und bei so niedriger Temperatur wie möglich (s.u.) in einem Nickel- oder Platintiegel(!) geschmolzen. Porzellantiegel sind ungeeignet, da das Reagens Aluminium aus dem Porzellan löst und die Analyse verfälschen kann. Die Reaktion ist beendet, sobald aus der klaren Schmelze Schwefeltrioxid als weißer Nebel zu entsteigen beginnt. Der Schmelzkuchen wird in verdünnter Schwefelsäure gelöst. Zum Aufschluss von Aluminiumoxid kann der Schmelzkuchen auch in verdünnter Natronlauge gelöst werden (Bildung von Tetrahydroxyaluminat). Der entstandene Komplex kann durch Säure als Aluminiumhydroxid gefällt werden.
Erklärung
[Bearbeiten]Das eigentliche Aufschlussreagens ist das in der Hitze entstehende Kaliumdisulfat und das sich daraus entwickelnde Schwefeltrioxid. Bei ca. 250 °C bildet sich zunächst aus Kaliumhydrogensulfat Kaliumdisulfat (Pyrosulfat):

Kaliumdisulfat zersetzt sich bei höheren Temperaturen in Kaliumsulfat und Schwefeltrioxid:

Aufschluß von Eisen(III)-oxid:

Da der alkalische Sturz nach der Ammoniumsulfidfällung Anfängern häufig Schwierigkeiten bereitet, kann für schwer nachweisbare Stoffe ein Kaliumhydroxidauszug aus der Ursubstanz hergestellt werden. Hierfür wird die Ursubstanz mit 3 Kaliumhydroxid-Plätzchen und 5 ml Wasser versetzt. Es fallen Kupfer, Bismut, Nickel, Cobalt, Eisen und Mangan unter Bildung schwerlöslicher Hydroxide aus. In Lösung verbleiben Antimon, Zinn, Aluminium, Zink und Chrom, die nun mit spezifischen Nachweisreaktionen nachgewiesen werden können.
Nachweisreaktionen
[Bearbeiten]Antimon
[Bearbeiten]Nachweis als Antimonsulfid
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Fällungsreaktion |
| pH | |
| Indikation | oranger Niederschlag |
Durchführung
[Bearbeiten]Hier wird konzentrierte Schwefelwasserstoffsäurelösung oder eine Alkalisulfidlösung zur Stoffprobe gegeben. Es muss jedoch beachtetet werden, dass vorher störende Arsen- und Zinn-Ionen auszufällen sind.
Erklärung
[Bearbeiten]- Antimon- und Sulfid-Ionen reagieren im wässrigen Milieu zu Antimon(III)-sulfid.

Eisennagelprobe
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Redoxreaktion |
| pH | <7 |
| Indikation | schwarze Flocken |
Durchführung
[Bearbeiten]Dazu wird ein Eisennagel in die Antimonsalz-Lösung gelegt. Es bilden sich nach einigen Minuten bis Stunden eine schwarze Schicht elementarem Antimon. Lässt man den Nagel jedoch über Nacht im Reagenzglas liegen, so scheidet sich alles mögliche, was noch in der Lösung schwimmt, ab. Beobachtet man jedoch eine schwarze Schicht, so kann diese im Sauren wieder gelöst werden und weiter untersucht werden, z.B. durch eine Sulfidfällung (siehe oben).
Erklärung
[Bearbeiten]- Antimon(III)-Ionen zu elementarem Antimon reduziert und elementares Eisen zu Eisen(II)-Ionen oxidiert.

Antimon-Nachweis mittels Marhscher Probe
[Bearbeiten]Alternativ kann auch die Marshsche Probe direkt aus der Ursubstanz durchgeführt werden.
Aluminium
[Bearbeiten]Aluminium kommt in der Ammoniumsulfidgruppe vor und fällt nach dem Alkalisturz als farbloses Hydroxid aus. Alternativ kann man auch einen Kaliumhydroxidauszug versuchen und das Aluminium dort fällen.
Nachweis als Cobaltaluminat
[Bearbeiten]- auch Cobaltblau, Dumonts Blau, Coelestinblau, Leithners Blau, Thénards Blau
| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Entstehung eines Mischphasenoxidpigments |
| pH | egal |
| Indikation | blaue Schmelze |
Durchführung
[Bearbeiten]Auf eine Magnesiarinne wird wenig Analysensubstanz und darauf ein paar Tropfen einer stark verdünnten Cobaltnitratlösung gegeben. Ist die Schmelze nach dem Glühen im Bunsenbrenner blau, war Aluminium in der Probe. Es ist auf eine geringe Dosierung des Cobaltnitrats zu achten, da bei Cobaltnitratüberschuss Cobaltoxid entsteht, das eine eventuelle Blaufärbung übertönen kann.
Erklärung
[Bearbeiten]- Aluminium-Ionen, Cobalt-Ionen und Oxidionen reagieren zum blauen Cobaltaluminat.

Cobaltaluminat ist ein Cobalt-Aluminium-Spinell, die Strukturformel lautet
- CoO·Al2O3 (Cobalt(II)-oxid und Aluminiumoxid)
Nachweis als fluoreszierender Morinfarblack
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Komplexbildung |
| pH | neutral bis essigsauer, vorher basisch |
| Indikation | grüne Fluoreszens mit UV-Licht |
Durchführung
[Bearbeiten]Zunächst mit Salzsäure versetzen, um eventuell vorhandenes Aluminium zu lösen. Anschließend mit Kaliumhydroxid stark alkalisch machen. Nun mit etwas Morin-Lösung versetzen und mit Eisessig (konzentrierte Essigsäure) ansäuern. Unter der UV-Lampe sollte die Lösung nun stark grün fluoreszieren. Hinweis: unbedingt Blindprobe machen und vergleichen, da Morin auch eine gewisse Eigenfluoreszenz hat.
Erklärung
[Bearbeiten]Al(III) bildet in neutralen sowie essigsauren Lösungen in Verbindung mit Morin eine fluoreszierende kolloidale Suspension.

Nachweis als Alizarin-S-Farblack
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Komplexbildung |
| pH | neutral bis essigsauer, vorher basisch |
| Indikation | Rotfärbung |
Eine saure Lösung mit Aluminium-Ionen wird mit möglichst wenig Kaliumhydroxid basisch gemacht und zentrifugiert. 1 Tropfen des Zentrifugats wird auf der Tüpfelpalette oder auf dem Objektträger mit 1 Tropfen 0,1%ige Natriumalizarinsulfonatlösung (Alizarin S) versetzt und 1 mol/l Essigsäure bis zum Verschwinden der rotvioletten Farbe und danach noch ein weiterer Tropfen Essigsäure zugegeben. Die Bildung eines roten Niederschlags oder eine Rotfärbung zeigt Aluminium an. Der Niederschlag wird häufig erst nach einigem Stehen sichtbar. Die rotgefärbte Verbindung ist in verdünnter Essigsäure schwer löslich, während die rotviolette Färbung der ammoniakalischen Alizarin-S-Lösung beim Ansäuern in Gelb umschlägt.
Störung
[Bearbeiten]Eisen, Chrom und Titan geben ähnlich gefärbte, gegen Essigsäure stabile Lacke. Auch Erdalkaliionen in konzentrierter Lösung geben gefärbte Niederschläge mit Alizarin, die jedoch in Essigsäure löslich sind.
Erklärung
[Bearbeiten]Aluminium-Ionen bilden mit dem Farbstoff Alizarin S einen sogenannten Farblack
- Im alkalischer Lösung bilden Aluminium-Ionen und Natriumalizarinsulfonat unter Abspaltung von Wasser einen Alizarin- Aluminium-Natriumkomplex.
![{\displaystyle {\begin{array}{ll}&\mathrm {[Al(OH)_{3}(H_{2}O)_{3}]+3\ C_{14}H_{7}SO_{7}Na} \\\longrightarrow &\mathrm {Na_{3}[Al(C_{14}H_{6}SO_{7})_{3}]\downarrow +\ 6\ H_{2}O} \end{array}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/59f1472dcd88247e4ddc6f1106d8c24da57632d1)

Arsen
[Bearbeiten]Arsen fällt in der Schwefelwasserstoffgruppe als gelbes Arsensulfid aus.
Nachweis mittels Marshscher Probe
[Bearbeiten]Man sollte unbedingt die Marshsche Probe ausprobieren. Auch wenn diese aus der Ursubstanz durchgeführt wird und wie eine Vorprobe wirkt, ist die Nachweisreaktion sehr spezifisch und kann auch kleinste Menge Arsen zuverlässig anzeigen.
Nachweis mittels Bettendorfsche Probe
[Bearbeiten]Nachweis mittels Gutzeitsche Probe
[Bearbeiten]Nachweis mittels Fleitmannsche Probe
[Bearbeiten]Arsenat-Nachweis mit Magnesiumsalz
[Bearbeiten]Arsenat-Ionen ähneln dem Phosphat-Anion. Entsprechend gibt es weitere, den Phosphat-Nachweisen ähnliche Reaktionen zur Identifikation von Arsenat:
Gibt man zu einer ammoniakalischen, ammoniumchloridhaltigen Lösung von Arsenat Magnesium-Ionen, so erhält man einen kristallinen Niederschlag von Magnesiumammoniumarsenat-Hexahydrat:
- Arsenat reagiert mit Magnesiumionen, Ammoniumionen und Wasser zu Magnesiumammoniumarsenat-Hexahydrat.

Arsenatnachweis mit Molybdatlösung
[Bearbeiten]Eine weitere, dem Phosphatnachweis ähnliche Nachweisreaktion von Arsenat in wässriger Lösung ist die Fällung mit Ammoniummolybdat. Der gelbe Niederschlag ist schwerlöslich in Säuren, aber gut löslich in Basen:
- Dihydrogenarsenat reagiert mit Wasserstoffionen, Ammoniumionen und Molybdationen zu Ammoniumarsenomolybdat und Wasser.
![{\displaystyle {\ce {H2AsO4- + 22H3O+ + 3NH4+ + 12MoO4^2- -> (NH4)3[As(Mo3O10)4*aq] +34H2O}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c4883783b577aaf6ddd2786047e84a878d6d04ef)
Bettendorfsche Probe
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Redoxreaktion |
| pH | <7 salzsauer |
| Indikation | schwarzer Niederschlag / Braunfärbung der Lösung |
Durchführung
[Bearbeiten]Bei der Bettendorfschen Probe wird die reduzierende Wirkung des Zinn(II)-chlorids ausgenutzt. Dieser Nachweis ist innerhalb der Arsengruppe spezifisch für Arsen. Es werden 5 Tropfen der Probelösung auf einem Uhrglas mit 3 Tropfen verdünntem Ammoniakwasser, 1 Tropfen 30%-igem Wasserstoffperoxid und 3 Tropfen 0,1 molarer Magnesiumchloridlösung versetzt und langsam zur Trockne eingedampft. Der Rückstand wird nach kurzem Erhitzen auf Rotglut mit 3 bis 5 Tropfen Zinn(II)-chloridlösung versetzt und schwach erwärmt. Ein schwarzer Niederschlag bzw. eine Braunfärbung der Lösung deutet auf Anwesenheit von Arsen. Sehr kleine Arsenmengen lassen sich nachweisen, wenn man mit Ether oder Amylalkohol ausschüttelt, die Folge ist eine schwarze Zone in der Grenzschicht.
Erklärung
[Bearbeiten]- Arsen(III)-Ionen reagieren mit Zinn(II)-Ionen und Chlorid-Ionen zu braunschwarzem, elementarem Arsen und dem Hexachlorostannat(IV)-Komplexion.
![{\displaystyle \mathrm {2\ As^{3+}+3\ Sn^{2+}+18\ Cl^{-}\longrightarrow 2\ As\downarrow +3\ [SnCl_{6}]^{2-}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/43e08c5fb68d8fde50cccef6b3245ad53e434c8b)
Nachweis von Arsen und Antimon mittels Fleitmannscher Probe
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Redoxreaktion |
| pH | >7 alkalisch |
| Indikation | Gelbfärbung, allmählich schwarz |
Bei der Fleitmannschen Probe wird im alkalischen Medium Arsenwasserstoff gebildet:
Durchführung
[Bearbeiten]Dazu wird die Probelösung in einem kleinen Erlenmeyerkolben mit Kaliumhydroxid und Aluminiumpulver erhitzt. Eventuell entstehender Schwefelwasserstoff wird mit Blei(II)-acetatlösung (auf einem Wattebausch in der Mündung des Reagenzglases) abgefangen. Die Öffnung des Kolbens wird mit einem Filterpapier bedeckt, das mit Silbernitratlösung oder Quecksilber(II)-chloridlösung getränkt ist. Eine Gelbfärbung, die allmählich in Schwarz übergeht bzw. eine sofortige Braunfärbung zeigen Arsen an.
Erklärung
[Bearbeiten]Reaktionsgleichungen für die Reaktion mit Silbernitrat sind identisch mit der Gutzeitschen Probe.
- Arsen(III)-Ionen reagieren mit Aluminium im alkalischen Medium zu Arsenwasserstoff und zum Tetrahydroxoaluminat(III)-Ion.
![{\displaystyle \mathrm {As^{3+}+3\ H_{2}O+5\ OH^{-}+2\ Al\longrightarrow AsH_{3}\uparrow +2\ [Al(OH)_{4}]^{-}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/9c0ce3bc952cdc02b6a92aec959775196c48d50e)
- Arsenwasserstoff reagiert mit Quechsilber(II)-chlorid zum braungefärbten Arsenmercurid und Chlorwasserstoff.

Nachweis von Arsen und Antimon mittels Gutzeitscher Probe
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Redoxreaktion |
| pH | <7 schwefelsauer |
| Indikation | Gelbfärbung, anschließende Schwärzung |
Durchführung
[Bearbeiten]Etwas Ursubstanz wird in einem kleinen Erlenmeyerkolben mit einer Zinkgranalie und etwas Schwefelsäure versetzt. Der Kolben wird mit einem Wattebausch verschlossen und auf seine Öffnung ein Filterpapier mit etwas festem Silbernitrat und einigen Tropfen Wasser gelegt. Durch Arsenwasserstoff kommt es zur Gelbfärbung des Nitrats und anschließender Schwärzung durch elementares Silber.
Erklärung
[Bearbeiten]Arsen(III)-Ionen bilden mit naszierendem Wasserstoff Arsenwasserstoff, welcher mit Silbernitrat zu dem gelben Doppelsalz Silberarsenidnitrat reagiert. Dieses färbt sich nach einigem Stehen unter Silberbildung schwarz.
- Arsenwasserstoff reagiert mit Silbernitrat zu Silberarsenidnitrat und Salpetersäure.

- Silberarsenidnitrat reagiert mit Wasser zu elementarem Silber, arseniger Säure und Salpetersäure.

Nachweis von Arsen und Antimon mittels Marshscher Probe
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Redoxreaktion |
| pH | <7 salzsauer |
| Indikation | orange Flamme, schwarzer Spiegel |

Die Marshsche Probe ist eine Nachweisreaktion, mit der man noch kleinste Mengen Arsen oder Antimon zuverlässig nachweisen kann. Sie stammt von dem englischen Chemiker James Marsh und erlangte 1832 Bekanntheit, weil es der erste gute gerichtsmedizinische Nachweis für das hochgiftige Arsen war. Es eignet sich gut als Vorprobe aus der Ursubstanz.
Durchführung
[Bearbeiten]Gefahren: Es gibt ein paar Dinge zu beachten: Arsensalze sowie das entstehende Arsenwasserstoffgas sind hochgiftig, also mit Handschuhen und unter dem Abzug arbeiten. Das Gasgemisch ist brisant, also vorher Knallgasprobe machen, ansonsten gibt es eine unangenehme kleine Explosion im Reagenzglas mit hochgiftigen Substanzen. Wenn man den Versuch aber richtig durchführt und sich an die Sicherheitsvorschriften hält, ist er nicht gefährlich.
Zunächst erwärmt man eine Tropfpipette, zieht diese vorsichtig aus, biegt sie wie in der Abbildung zurecht und steckt das dickere Ende der Pipette durch einen Stopfen. Die Konstruktion kann mehrfach verwendet werden. Man gibt etwas Salzsäure, Zinkperlen, Kupfersulfat und einen Teil der Analysensubstanz in ein Reagenzglas (1). Es sollte eine sprudelnde Reaktion unter Bildung von Wasserstoffgas entstehen. Bevor man weitermacht, sollte man eine Knallgasprobe machen: Wenn es nur noch leicht ploppt, setzt man die präparierte Pipette mit Stopfen auf. Man kann jetzt einen Teil des Glasrohres erwärmen (2) und im Glas auf einen Metallspiegel achten (3). Meist zündet man jedoch den Wasserstoff an dem verjüngten Ende Pipette an (4) und kann dann mit der Flamme auf der Innenseite einer Porzellanabdampfschale einen schwarzen Spiegel hinterlassen. Man kann damit regelrecht „malen“.
Um nun zu prüfen, ob Antimon oder Arsen in der Probe ist, gibt man eine ammoniakalische Wasserstoffperoxid-Lösung auf den schwarzen Spiegel. Arsen löst sich, Antimon nicht. Man kann den schwarzen Spiegel auch weiter untersuchen, um zweifelsfrei Arsen nachzuweisen. Dafür erhitzt man die Lösung, um Wasserstoffperoxid zu vertreiben, säuert an und versucht eine Fällung mit Schwefelwasserstoff. Arsen fällt dabei als gelbes Arsensulfid und kann weiter untersucht werden.
Erklärung
[Bearbeiten]Der Nachweis beruht darauf, dass Zink und Säure naszierenden (sehr reaktiven) Wasserstoff bilden, der sofort mit Arsenik den Arsenwasserstoff bildet. Das Kupfersulfat dient dazu, die Reaktion durch Bildung eines Lokalelements an den Zinkstückchen zu beschleunigen.
- Arsen(III)-oxid reagiert mit Zink in saurer Lösung zu Arsenwasserstoff, Zink(II) und Wasser.

Barium
[Bearbeiten]Barium verbleibt im Trennungsgang in der Ammoniumcarbonatgruppe. Es lässt sich wie die meisten Erdalkaliionen bevorzugt mittels Flammenfärbung aufspüren.
Nachweis als Bariumsulfat
[Bearbeiten]_reactions.JPG)
| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Fällungsreaktion |
| pH | egal |
| Indikation | weißer Niederschlag |
Durchführung
[Bearbeiten]Zur Probelösung wird Sulfatlösung (z.B. verdünnte Schwefelsäure) geben. Mit Ba2+ entsteht ein weißer Niederschlag.
Erklärung
[Bearbeiten]- Bariumkationen und Sulfatanionen reagieren zum weißen Bariumsulfat.

Störung
[Bearbeiten]Bismut
[Bearbeiten]Nachweis als Bismutiodid
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Fällungsreaktion, Komplexbildung |
| pH | <7 HNO3 oder H2SO4 |
| Indikation | schwarzer Niederschlag, orange Lösung |
Durchführung
[Bearbeiten]Nach Zugabe von Iodidlösung fällt schwarzes Bismutiodid aus, welches sich im Iodidüberschuss als oranger Komplex löst.
Erklärung
[Bearbeiten]Zunächst eine Fällungsreaktion zu schwarzem Bismutiodid:
- Bismut(III)-Ionen und Iodid-Ionen reagieren zu Bismut(III)-iodid.

Im Überschuss von Iodidlösung kommt es zur Bildung eines orangen Komplexes:
- Bismut(III)-iodid und Iodid-Ionen reagieren zum Tetraiodobismutat(III)-Komplex.
![{\displaystyle \mathrm {BiI_{3}+I^{-}\longrightarrow [Bi(I)_{4}]^{-}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/10a6412305dcdcfb273d2bdafefa7bc80a643ab3)
Nachweis als Bismut
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Redoxreaktion |
| pH | 6,5-8 |
| Indikation | schwarzer Niederschlag |
Durchführung
[Bearbeiten]Zum Nachweis von Bismut(III)-Kationen sollte die zu untersuchende Lösung erst einmal neutralisiert werden (pH 6,5-8). Anschließend wird alkalische Zinn(II)-Lösung hinzugegeben. Die Zinn(II)-Ionen wirken dabei als Reduktionsmittel, sie reduzieren also Bismut(III)-Ionen zu elementarem, schwarzem Bismut, welches in wässriger Lösung ausfällt.
Erklärung
[Bearbeiten]- Bismut(III)-Ionen und Zinn(II)-Ionen reagieren zu elementarem Bismut und Zinn(IV)-Ionen.

Nachweis mittels Bismutrutsche
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Komplexbildungsreaktion |
| pH | <7 HNO3 |
| Indikation | zitronengelber Komplex |
Durchführung
[Bearbeiten]
Zum Nachweis aus der Ursubstanz kann man die "Bismutrutsche" mit Thioharnstoff verwenden. Dieser Nachweis gilt als ziemlich sicher, da eventuell störende Ionen vorher ausfallen. Ein angefeuchtetes Filterpapier wird in der Mitte geknickt und in folgender Reihenfolge mit der Ursubstanz und den Fällungsmitteln beschichtet. Zunächst legt man die Ursubstanz auf das Filterpapier, dann Natriumfluorid, welches mit Aluminium und Eisen einen Komplex bildet, nun Natriumchlorid, welches Silber und Quecksilber fällt, es folgt Kaliumnatriumtartrat, das mit Antimon und Zinn einen Komplex bildet und schließlich Thioharnstoff als eigentliche Nachweisreagenz. Nun hält man das Filterpapier schräg und tropft verdünnte Salpetersäure darauf und lässt diese auf dem Filterpapier "rutschen". Bei Anwesenheit von Bismut entsteht ein zitronengelber Thioharnstoff-Komplex.
Erklärung
[Bearbeiten]![{\displaystyle \mathrm {Bi^{3+}+3\ SC(NH_{2})_{2}\longrightarrow [Bi(SC(NH_{2})_{2})_{3}]^{3+}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/965449e8be3b503fde7a382cb4541b0160ed7321)
Blei
[Bearbeiten]Nachweis als Bleiiodid
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Fällungsreaktion |
| pH | <7 bis 7 |
| Indikation | gelber Niederschlag |
Durchführung
[Bearbeiten]Gibt man zu der Analysenlösung etwas Kaliumiodid, so entsteht ein voluminöser gelber Niederschlag, der sich im Überschuss von Iodidlösung wieder löst.
Die Verbindung lässt sich auch Umkristallisieren: dafür erhitzt man die Lösung mit einem Bunsenbrenner, so dass das Blei(II)-iodid wieder in Lösung geht. Nach dem Abkühlen fällt das Bleiiodid wieder in gelben glitzernden Plättchen aus.
Erklärung
[Bearbeiten]- Blei(II)-Ionen und Iodid-Ionen reagieren zum schwer löslichen, gelbfarbenden Blei(II)-iodid.

Nach Zugabe weiterer Iodidlösung reagiert das Blei(II)-Iodid zum farblosen Tetraiodoplumbat(II)-komplex weiter.
- Blei(II)-iodid und Iodid-Ionen reagieren zum gut löslichen, farblosen Tetraiodoplumbat(II)-komplex.
![{\displaystyle \mathrm {PbI_{2}+2\ I^{-}\longrightarrow [Pb(I)_{4}]^{2-}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/e7179e755ef4d8e6d9bbdecddc45509d3ccb12b9)
Störung
[Bearbeiten]Der Nachweis wird durch viele andere Schwermetall-Kationen gestört, daher müssen diese vorher abgetrennt werden. Im Kationentrennungsgang erfolgt dies in der Salzsäuregruppe und in der Schwefelwasserstoffgruppe.
Nachweis als Bleichromat
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Fällungsreaktion |
| pH | >7 |
| Indikation | gelber Niederschlag, in Natronlauge rot |
Durchführung
[Bearbeiten]Bei Versetzen einer Blei(II)-Ionen-haltigen Lösung mit Kaliumchromat kommt es zur Bildung eines gelben, in Essigsäure und Ammoniak unlöslichen, jedoch in Natronlauge und Salpetersäure löslichen, kristallinen Niederschlages. Die Kristallstruktur kann unter dem Mikroskop betrachtet werden. Dabei ist zu beachten, dass die Reaktion aufgrund des "Chromat-Dichromat-Gleichgewichtes" im richtigen pH-Wert-Bereich (am besten >6) durchgeführt wird.
Die schwach alkalische Lösung wird mit wenig verdünnter Kaliumchromat-Lösung versetzt und anschließend mit verdünnter Essigsäure schwach angesäuert, Folge: ein Niederschlag von gelbem, schwer löslichem Bleichromat, das sich in Essigsäure nicht löst, entsteht. Beim Behandeln mit etwas Natronlauge bildet sich rotes, basisches Bleichromat.
Erklärung
[Bearbeiten]- Blei(II)-Ionen und Chromat-Ionen reagieren zum schwer löslichen, gelbem Bleichromat.

- Bleichromat reagiert mit Natronlauge zu rotbraunem basischem Bleichromat und Natriumchromat.

Bor
[Bearbeiten]Bor kommt im anorganischen Praktikum als Borat BO33– oder Borsäure H3BO3 vor.
Borat
[Bearbeiten]Die Standardreagenz dieser Stoffklasse ist Borax (Natriumborat), ein weißes bis gräulich gefärbtes Salz.
Borat-Nachweis als Methylester
[Bearbeiten]Durchführung
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Veresterung |
| pH | <7 schwefelsauer |
| Indikation | grüne Flamme |

Die Analysensubstanz wird mit Methanol und einigen Tropfen Schwefelsäure als Katalysator versetzt. Die Dämpfe werden in einem abgedunkelten Abzug vorsichtig angezündet. Der Nachweis ist positiv, wenn sich die Flamme grün färbt.
Erklärung
[Bearbeiten]- Borat-Ionen und Methanol reagieren unter dem Katalysator Schwefelsäure zu Trimethylborat und Hydroxid-Ionen.

Die Dämpfe des Methanol/Trimethylborat-Gemisches werden entzündet. Es erscheint eine leuchtend grüne Flamme.
- Beim Verbrennen des Trimethylborats entsteht Kohlenstoffdioxid, Wasser und Bortrioxid

Borsäure
[Bearbeiten]Borsäure (Orthoborsäure) ist ein weißer Feststoff.
Mannito-Borsäure-Komplex
[Bearbeiten]Durchführung
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Komplexbildung |
| pH | =7 neutral |
| Indikation | Absinken des pH-Wertes |
Ein wässriger Auszug wird mit 0,1 molarer Natronlauge auf einen pH-Wert von 7 eingestellt. Nach der Zugabe von 0,5 g Mannit wird der pH-Wert erneut vermessen. Ein deutliches Absinken des pH-Wertes zeigte die Anwesenheit von Borsäure in der Probe an.
Erklärung
[Bearbeiten]Die sehr schwache Säure wird durch Umsetzung mit mehrwertigen Alkoholen wird in ihrer Säurestärke erheblich gesteigert:

Dies ist bedingt durch eine Verschiebung des Gleichgewichtes auf die rechte Seite hin zu einem Tetraoxoborat-Derivat in Folge einer Veresterung.
Cadmium
[Bearbeiten]Nachweis als Cadmiumsulfid
[Bearbeiten]Cadmium weist man durch Zugabe von Natrium- oder Ammoniumsulfidlösung zur essigsauren Cadmiumsalzlösung nach: Es entsteht ein gelber Niederschlag von Cadmiumsulfid. Im Kationentrenngang ist Cadmium zuvor von störenden Begleitmetallen bzw. -schwermetallkationen im Schwefelwasserstoffgruppe zu trennen. Insbesondere Kupferionen stören hier diesen Nachweis und müssen zuvor mit giftigem Kaliumcyanid „maskiert“ werden.
Durchführung
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Fällungsreaktion |
| pH | >7 |
| Indikation | gelber Niederschlag |
Kupfersalze müssen im Kationentrenngang vor dem Cadmiumnachweis aufwändig in einen farblosen Tetracyanidocuprat(II)-Komplex überführt werden: Nach Zugabe von Zyankali (KCN) zur Hauptlösung der Kupfergruppe muss sich die Lösung entfärben (ein zusätzlicher Nachweis für Cu; Achtung: Ab hier die Lösung nicht mehr ansäuern, sonst entsteht hochgiftige Blausäure (HCN-Gas)! Bei der Entsorgung beachten – mit konz. Wasserstoffperoxid entgiften!). Wenn man bis zur vollständigen Entfärung KCN zugegeben hat, kann man dann mit einer Sulfid-Lösung das gelbe Cadmiumsulfid CdS ausfällen, ohne dass schwarzes Kupfer(II)-sulfid stört.
Erklärung
[Bearbeiten]- Kupfer und Cyanid reagiert zum farblosen Tetracyanidocuprat(II)-Komplex
![{\displaystyle \mathrm {Cu^{2+}+4\ KCN\longrightarrow [Cu(CN)_{4}]^{2-}+4\ K^{+}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/ac65d3f081d92afe3982a874384001136fe77b23)
- Cadmium(II)-Ionen und Sulfid-Ionen reagieren im wässrigen Milieu zum gelben Cadmiumsulfid, welches ausfällt.

Carbonat
[Bearbeiten]Carbonat-Nachweis nach Kohlendioxidentwicklung
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Verdrängungsreaktion |
| pH | <7 salzsauer |
| Indikation | getrübtes Barytwasser |

Durchführung
[Bearbeiten]Carbonat-Ionen (CO32−) lassen sich durch Zugabe von Salzsäure nachweisen, bei der Kohlenstoffdioxid entsteht. Als Vorversuch kann man festes Analysegut mit konzentrierter Salzsäure versetzen. Eine Gasentwicklung (CO2) deutet auf Carbonat hin. Bei Durchführung dieses Tests im Reagenzglas sollte die Flamme eines eingebrachten brennenden Spans erstickt werden.
Das entstehende Gas kann auch durch eine Fällungsreaktion identifiziert werden: Das gasförmige Kohlenstoffdioxid wird in Kalk- oder Barytwasser geleitet, z. B. über eine Rohrkonstruktion. Einfacher ist es, ein Gärröhrchen (Carbonatröhrchen), das an dem einen Ende in einem durchbohrten Stopfen steckt, mit Kalk- oder Barytwasser zu füllen und mit Stopfen auf das Reagenzglas mit Säure und Analysensubstanz zu stecken. Der Nachweis ist positiv wenn sich eine weiße Trübung von Calcium- bzw. Bariumcarbonat bildet.
Falls man Blasen sieht, aber die Kalk-/Barytwasserlösungen sich nicht trüben, kann es sein, dass das Carbonat zu schnell ausgetrieben wird. Dann bietet es sich an, eine schwächere Säure zu nehmen (z. B. Essigsäure) und das Gemisch länger im Wasserbad mit Gärröhrchen zu erwärmen. Die Trübung bildet sich dann mit der Zeit.
Erklärung
[Bearbeiten]- Carbonate reagieren mit Salzsäure zu gasförmigem Kohlendioxid, Chlorid und Wasser

- Bariumhydroxid und Kohlenstoffdioxid reagieren zu Bariumcarbonat und Wasser

Störung
[Bearbeiten]Bei dem Versuch ist der störende Einfluss von Sulfit- und Thiosulfationen zu beachten. Diese können durch vorheriges Zutropfen von Wasserstoffperoxidlösung entfernt werden.
Chrom
[Bearbeiten]Chrom kommt in den klassischen Trennungsgängen in der Ammoniumsulfidgruppe im alkalischen Sturz vor und kann auch im Kaliumhydroxidauszug abgetrennt werden.
Nachweis als Chromat
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Redoxreaktion |
| pH | >7 |
| Indikation | gelbe Lösung (in Säuren orange), mit Barium gelber NS |
Chrom(III)-salze ergeben bei der Reaktion mit dem „Alkalischen Bad“ (Ätznatron in konz. Wasserstoffperoxidlösung) gelbe Chromatlösungen, die im Sauren zu orangem Dichromat reagieren:
- Chrom(III)-Ionen reagieren im alkalischen Bad zu gelben Chromat-Ionen und Wasser.

Auch im sauren Medium können Chrom(III)-salze aufoxidiert werden. Dazu sind jedoch besonders starke Oxidationsmittel notwendig, so zum Beispiel Peroxodisulfate.
- Chrom(III)-Ionen reagieren im wässrigen Milieu mit Peroxodisulfaten zu orangefarbenden Dichromat-Ionen sowie Sulfat-Ionen und Wasserstoff-Ionen.

Bei der Oxidationsschmelze mit Soda und Salpeter werden Chrom(III)-Ionen hingegen wieder zu gelben Chromat-Ionen gemäß folgender Reaktionsgleichung aufoxidiert:
- Chrom(III)oxid reagiert mit Soda und Salpeter zu gelbfarbendem Natriumchromat, Kaliumnitrit und Kohlenstoffdioxid.

Auch bei den Vorproben tritt beim Schmelzen der Salzperle mit Phosphorsalz oder Borax eine charakteristisch grüne Färbung mit Chrom(III)-Ionen auf.
Mit Bariumsalzen entsteht ein gelbes Bariumchromat. Hierfür wird alkalische Chromatlösung mit Eisessig sauer gemacht (pH 3-5) und mit Natriumacetat gepuffert. Anschließend kann mit Bariumchlorid ausgefällt werden. Bariumchromat löst sich in Salzsäure wieder.
- Bariumsalzen fällen Chromate als gelbes Bariumchromat

Nachweis als Chrompentoxid
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Komplexbildung |
| pH | <7 |
| Indikation | blaue Etherschicht (oben) |
Durchführung
[Bearbeiten]Die Lösung muss salpetersauer sein. Sie wird im Reagenzglas mit Ether überschichtet, mit einigen Tropfen Wasserstoffperoxid (30%) versetzt und geschüttelt. Bei einer blauen Etherschicht (oben) war Chrom vorhanden. Die Färbung verschwindet bald wieder und ist wenn überhaupt dann nur leicht blau.

Erklärung
[Bearbeiten]Der blaue Komplex von Chrompentoxid (auch Chrom(VI)-peroxid oder Schmetterlingskomplex) ist nur in Ether stabil.
_reactions.JPG)
Cobalt
[Bearbeiten]Cobalt-Kationen werden im Kationentrennungsgang in der Ammoniumsulfidgruppe neben Nickel-Kationen als schwarzes Cobalt(II)-sulfid gefällt.
- Cobalt-Kationen reagieren in nichtsaurer, acetathaltiger Lösung mit Ammoniumsulfid zum schwarzem Cobalt(II)-sulfid und Ammonium-Ionen.

Wird unter starkem Luftzutritt und bei Gegenwart von überschüssigem Ammoniumsulfid gefällt, bildet sich aus Cobalt(II)-sulfid zunächst Hydroxocobalt(III)-sulfid, das in Cobalt(III)-sulfid übergeht.
- Cobaltsulfid reagiert in wässriger Lösung mit Sauerstoff zum Hydroxocobalt(III)-sulfid, welches mit Schwefelwasserstoff zum Cobalt(III)-sulfid weiterreagiert. Als Nebenprodukt entsteht Wasser.
![{\displaystyle \mathrm {4\ CoS+O_{2}+2\ H_{2}O\longrightarrow 4[Co(OH)]S} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/96adb974991718d38c6faaf09b181de7d1b0e3d5)
![{\displaystyle \mathrm {2\ [Co(OH)]S+H_{2}S\longrightarrow Co_{2}S_{3}+2\ H_{2}O} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/1b4cc742fc1c510d43097de5c2837433df19c454)
Eine relativ aussagekräftige Vorprobe für Cobalt ist die Borax- und Phosphorsalzperle, die von Cobaltionen intensiv blau gefärbt wird.
Nachweis als Cobalthydroxid
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Komplexbildung |
| pH | >7 |
| Indikation | blauer, im Überschuss roter Niederschlag |
Bei Zugabe einer starken Hydroxidlösung, z. B. Natriumhydroxidlösung zu der zu untersuchenden Stoffprobe bildet sich zuerst ein blauer Niederschlag eines basischen Cobaltsalzes wechselnder Zusammensetzung.
- Cobalt-Kationen reagieren in der Kälte (max. 15°C) mit Hydroxid-Ionen zum Hydroxocobalt(II)-komplexion. Der Komplex kann mit verschiedensten Anionen basische Salze bilden.
![{\displaystyle \mathrm {Co^{2+}+OH^{-}\longrightarrow [Co(OH)]^{+}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/d31ad908747e3e633966cb4290ed76fd74855b27)
Bei Erhitzung der immer noch alkalischen Probelösung zerfällt das Hydroxocobalt(II)-komplexion und es bildet sich das rosenrote Cobalt(II)hydroxid.
- Hydroxocobalt(II)-Ionen reagieren unter Hitze in alkalischer Lösung in das rosenrote Cobalt(II)hydroxid.
![{\displaystyle \mathrm {[Co(OH)]^{+}+OH^{-}\longrightarrow Co(OH)_{2}\downarrow } }](https://wikimedia.org/api/rest_v1/media/math/render/svg/452dd8bca0fcfb64e02ff14a05b2eaa47010138c)
Nachweis als Thiocyanatokomplex
[Bearbeiten]
| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Komplexbildung |
| pH | |
| Indikation | pinke Lösung |
Bei Zugabe von Thiocyanat (am besten festes Salz statt Lösung) entsteht je nach Konzentration eine pinkfarbene bis blaue Lösung. Der blaue Komplex kann mit einem organischen Lösungsmittel (z. B. Butanol) extrahiert werden.
Erklärung
[Bearbeiten]- Cobalt-Kationen reagieren im wässrigen Milieu bei Zugabe von Thiocyanat-Ionen zum pinken Pentaaquathiocyanatocobalt(II)-komplex.
![{\displaystyle \mathrm {Co^{2+}+SCN^{-}+5\ H_{2}O\longrightarrow [Co(H_{2}O)_{5}(SCN)]^{+}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/96edafdb26bd782483e7a66e128e25544361bb58)
Störung
[Bearbeiten]Der Nachweis mit Thiocyanat interferiert mit Eisen- und Kupfer-Kationen.
Eisen
[Bearbeiten]Eisen lässt sich mit Blutlaugensalzen aus der Ursubstanz nachweisen, falls kein weiterer Stoff mit dem Blutlaugensalz ebenfalls zu farbigen Niederschlägen reagiert, siehe Nachweisreagenzien#Blutlaugensalze.
Im Kationentrennungsgang fällt Eisen erstmals in der Hydrolysegruppe als braunes Eisenhydroxid: Zu dem Filtrat aus der Schwefelwasserstoffgruppe gibt man konzentrierte Salpetersäure um Fe2+ zu Fe3+ zu oxidieren. Dann gibt man dazu noch konzentriertes Ammoniakwasser.
- Eisen(III)-Kationen reagieren mit Ammoniak zu braunem Eisenhydroxid und Ammoniumionen

Danach erfolgt die Abtrennung in der Ammoniumsulfidgruppe:
- Eisenhydroxid und Ammoniumsulfid reagiert zu braunem Eisen(II)-sulfid und Ammoniumhydroxid

Eisen(II) mit rotem Blutlaugensalz
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Bildung eines Charge-Transfer-Komplexes |
| pH | <7 salzsauer |
| Indikation | tiefblauer Niederschlag |
,(III).JPG)
Hierfür wird die Analysenlösung mit verdünnter Salzsäure und rotem Blutlaugensalz versetzt. Dabei entsteht ein Eisen(II)-Eisen(III)-Komplex, der tiefblau ist und sich in wässriger Lösung langsam absetzt, er ist also schwer wasserlöslich (siehe Bild zweites Reagenzglas von links). Das Pigment trägt den Namen Berliner Blau (auch Pariser Blau, Französischblau, Eisencyanblau, Turnbulls Blau, Bronzeblau, Preußisch Blau, Pottascheblau, Chinesischblau, Miloriblau, Stahlblau, Tintenblau, Tonerblau).
Erklärung
[Bearbeiten]Es läuft in gewissem Sinne jedoch keine Komplexbildungsreaktion ab, sondern zunächst lediglich ein Ionenaustausch / Fällungsreaktion, in dessen Niederschlagsprodukt dann jedoch beide Eisenionen unterschiedlicher Wertigkeit wie in einem agieren können (engl.: charge transfer):
- Eisen(II)-Ionen reagieren mit Kaliumhexacynanidoferrat(III) zu einem Eisenhexacyanidoferratkomplex und Kaliumionen
\longrightarrow Fe_{3}[Fe(CN)_{6}]_{2}+6\ K^{+}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/1f3fcbb832458896fe5d5765428e3e4abc807d25)
Eisen(III) mit gelbem Blutlaugensalz
[Bearbeiten]Durchführung
[Bearbeiten]Eisen(III)-Ionen lassen sich analog mit gelbem Blutlaugensalz nachweisen, wobei ein Eisen(III)-Eisen(II)-Komplex entsteht, der auch tiefblau gefärbt ist, aber im Gegensatz zum Eisen(II)-Eisen(III)-Komplex sich kolloid in Wasser löst.
Erklärung
[Bearbeiten]- Eisen(III)-Ionen reagieren mit Kaliumhexacyanidoferrat(II) zu einem Eisenhexacyanidoferratkomplex und Kaliumionen.
\longrightarrow Fe_{4}[Fe(CN)_{6}]_{3}+12\ K^{+}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/cb616607c4333f7263b2800fa070cf198aff2b07)
Bei dieser Nachweisreaktion entsteht Berliner Blau, ein wichtiger Farbstoff. Turnbulls Blau und Berliner Blau sind trotz der hier angegebenen, unterschiedlichen Formeln identisch – nur ihre Herstellungswege sind unterschiedlich: Die Gewinnung von Turnbulls Blau erfolgt durch das Umsetzen von Eisen(II)-salzen mit Kaliumhexacyanoferrat(III) (rotem Blutlaugensalz) in wässriger Lösung – die von Berliner Blau durch Umsetzen von Eisen(III)-salzen mit Kaliumhexacyanoferrat(II) (gelbes Blutlaugensalz).
Mittels EPR- und Mößbauerspektroskopie konnte jedoch festgestellt werden, dass die Reaktionsprodukte beider Nachweisreaktionen weitgehend identisch sind, da folgendes Gleichgewicht besteht:
![{\displaystyle \mathrm {Fe^{2+}+[Fe(CN)_{6}]^{3-}\ \rightleftharpoons \ Fe^{3+}+[Fe(CN)_{6}]^{4-}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/ba48a61dbef287bc9886697f83bbae98997a0c18)
Eisen(III) mit Thiocyanat
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Komplexbildung |
| pH | <7 salzsauer |
| Indikation | blutroter Niederschlag |
-Ionen_und_Thiocyanat.JPG)
Durchführung
[Bearbeiten]Alternativ kann man Eisen(III)-salze (siehe Bild linkes Reagenzglas, hier als Beispiel Eisen(III)-chlorid) einsetzen. Die Analysenlösung wird mit verdünnter Salzsäure versetzt und in dieser kann dann durch Zugabe einer Thiocyanatlösung Eisen nachgewiesen werden. Als Reagenzien eignen sich Kaliumthiocyanat oder Ammoniumthiocyanat („Rhodanid“). Dieser Nachweis wird manchmal wegen der Färbung bei positiver Probe auch als „Stierblutprobe“ bezeichnet.
Die Probe ist sehr empfindlich und wird auch in der Spurenanalytik eingesetzt. Hier ist besonders sauberes Arbeiten nötig, um das Ergebnis nicht zu verfälschen (z. B. keinen Edelstahlspatel in die salzsaure Probelösung eintauchen). Sicherheitshalber auch eine Blindprobe durchführen um eine Verunreinigung der Salzsäure oder des Reagens' selbst durch Eisen, z. B. aus Rost, welcher in kleinsten Partikeln in der Luft vorhanden sein könnte, und somit ein falsch-positives Ergebnis, auszuschließen.
Erklärung
[Bearbeiten]Es reagieren dabei die Eisen(III)-Kationen mit den Thiocyanat-Ionen zu einem blutroten Komplex, dem Pentaaquathiocyanatoeisen(III)-Ion. (siehe Bild rechtes Reagenzglas)
- Eisen(III)-Ionen und Thiocyanat-Ionen reagieren in einem wässrigen Milieu zum Pentaaquathiocyanatoferrat(III)-komplex.
![{\displaystyle \mathrm {Fe^{3+}+\ SCN^{-}+5\ H_{2}O\longrightarrow [Fe(SCN)(H_{2}O)_{5}]^{2+}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/b06dd1440c76583922289bb01afc8baeac9e56ed)
Halogenide
[Bearbeiten]Halogenide sind chemischen Verbindungen der 7. Hauptgruppe des PSE (Halogene) mit der Oxidationszahl -I und kommen in Salzen als einfach negativ geladene Ionen daher.
Nachweise mit Silbersalzlösung
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Fällungsreaktion |
| pH | <7 salpetersauer |
| Indikation | weiße Niederschläge |
Die Silbersalze der Halogenide Chlorid, Bromid und Iodid zeichnen sich durch ihre Schwerlöslichkeit aus. Aus salpetersaurer Lösung fallen sie als dicker („käsiger“) weißer (Chlorid) bzw. gelblicher (Bromid, Iodid) Niederschlag aus.
Zu beachten ist, dass Fluorid-Ionen, die ja auch unter die Halogenid-Ionen zählen, keinen Niederschlag mit Silbersalzlösung bilden, da Silberfluorid ein in Wasser gut lösliches Salz ist.
Durchführung
[Bearbeiten]
Zum Nachweis wird die Probelösung mit Salpetersäure HNO3 angesäuert und mit etwas Silbernitratlösung AgNO3 versetzt. Der Niederschlag von Silberchlorid AgCl ist in Ammoniumcarbonatlösung (NH4)2CO3 löslich, wobei der Diamminsilber(I)-chlorid-Komplex [Ag(NH3)2]Cl entsteht. Der Niederschlag von Silberbromid AgBr löst sich in konzentrierter Ammoniaklösung NH3 aq, und der von Silberiodid AgI bleibt zurück.
Erklärung
[Bearbeiten]Bei Iodid-Ionen (siehe Bild Reagenzglas 1): Ausbildung eines käsig-gelben Niederschlags. Silberiodid ist gänzlich unlöslich in Ammoniakwasser.
- Iodid-Ionen reagieren mit Silbernitrat zu Silberiodid und Nitrat-Ionen.

Bei Bromid-Ionen (siehe Bild Reagenzglas 3): Ausbildung eines weiß/gelblichen Niederschlags.
- Bromid-Ionen reagieren mit Silbernitrat zu Silberbromid und Nitrat-Ionen.

Silberbromid ist in konz. Ammoniakwasser etwas löslich. (Reagenzglas 4)
- Silberbromid reagiert mit Ammoniakwasser zum löslichen Silberdiammin-Komplexion und Bromid-Ionen.
![{\displaystyle \mathrm {AgBr+2\ NH_{3}\longrightarrow [Ag(NH_{3})_{2}]^{+}+Br^{-}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/2fa4414adb1733f84ca605cc34ad7faeb77e99ff)
Bei Chlorid-Ionen (siehe Bild Reagenzglas 5): Ausbildung eines weißen Niederschlags.
- Chlorid-Ionen reagieren mit Silbernitrat zu Silberchlorid und Nitrat-Ionen.

Silberchlorid ist selbst in schwach konz. Ammoniakwasser recht gut löslich. (Reagenzglas 6)
- Silberchlorid reagiert mit Ammoniakwasser zum löslichen Silberdiammin-Komplexion und Chlorid-Ionen.
![{\displaystyle \mathrm {AgCl+2\ NH_{3}\longrightarrow [Ag(NH_{3})_{2}]^{+}+Cl^{-}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/366720889375d553e2cb96d4a5b72beaff839b02)
-
 AgCl (links); +NH3 (rechts)
AgCl (links); +NH3 (rechts) -
 AgBr (links); +NH3 (rechts)
AgBr (links); +NH3 (rechts) -
 AgI (links); +NH3 (rechts)
AgI (links); +NH3 (rechts)


Störung
[Bearbeiten]Wenn man das Filtrat mit verdünnter Salpetersäure ansäuert, sollte das störende Ausfallen von Silbercarbonat vor der Zugabe von Silbernitratlösung verhindert werden.
Vor der Analyse sollte eine Sodaauszug vorgenommen werden, da z. B. Kupferionen stören, weil eine Kupfer(II)-salzlösung bei Zugabe von Ammoniak aufgrund der Bildung des Amminkomplexes [Cu(NH3)4]2+ tiefblau wird.
Silberhalogenidfällungen mit Zink unterscheiden
[Bearbeiten]Gibt man zu dem in Ammoniak gelösten Silberbromid elementarem Zink (Zn), so wird das Silber reduziert und somit Br− in der Lösung freigesetzt. Dieses lässt sich nun mit Chlorwasser über die braune Färbung nachweisen.
Auch der Silberiodid-Niederschlag kann mit Zn reduziert werden, wobei die freiwerdenden Iodid-Ionen in Lösung gehen können. Auch dieses kann durch versetzen mit Chlorwasser nachgewiesen werden (violette Färbung)
| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Redoxreaktion |
| pH | <8 |
| Indikation | Brom: orangebraun Iod: rosaviolett, in O-haltigen LöMi braun |
Nachweis als Brom und Iod
[Bearbeiten]Eine weitere Unterscheidungsmöglichkeit für Bromid und Iodid ist die Zugabe von Chlorwasser oder (wenig) Chloramin T mit Salzsäure wobei die Halogenidionen zum Halogen oxidiert werden. Man gibt dann ein organisches Lösungsmittel wie n-Hexan hinzu und löst die Halogene durch kräftiges Schütteln in der organischen Phase ( Extraktion). Brom oder Iod kann dort einfach aufgrund der Färbung identifiziert werden. Alternativ kann man auch mit Chloroform extrahieren. Die organische Phase färbt sich braun bei Bromid und violett bei Iodid. Liegen beide Elemente vor, so ergibt sich eine Mischfarbe. Mit einiger Übung kann hier der Anteil der beiden Halogenide an der Mischung abgeschätzt werden. In sauerstoffhaltigen Lösungsmitteln wie Diethylether ist Iod braun.
Erklärung
[Bearbeiten]Löst man Chloramin T in Wasser so entsteht Hypochlorit, welches wiederum in Salzsäure nicht stabil ist.
- Hypochlorit mit Salzsäure synproportioniert zu Chlor und Chlorid.

Aufgrund der elektrochemischen Spannungsreihe entsteht zunächst Iod und anschließend Brom.
- Iodid-Ionen werden durch Chlor zu elementarem Iod oxidiert, welches sich in Hexan mit einer dunkelvioletten Färbung löst.
- Bromid-Ionen werden durch Chlor zu elementarem Brom oxidiert, welches sich in Hexan mit einer braunorangenen Färbung löst.


Die Folgereaktion zu Bromchlorid färbt die Lösung weingelb.
- Brom und Chlor reagieren zum weingelbem Bromchlorid

Störung
[Bearbeiten]Gestört wird dieser Nachweis, wenn die zu untersuchende Lösung gleichzeitig Sulfid- oder Thiosulfat-Anionen enthält. In diesem Fall fällt gelbweißer Schwefel als Trübung aus, da das Chlorwasser Sulfid zu Schwefel oxidiert. Auch darf der pH-Wert nicht über 8 liegen, da Halogene im alkalischen zu Hypohalogenitionen reagieren.
Im Überschuss von Chloramin T kann die Lösung farblos werden, daher am besten nur eine 10-prozentige Lösung ansetzen.
Brom
[Bearbeiten]Brom fällt in anorganischen Salzen als Bromid- und Bromatanion an.
Bromid
[Bearbeiten]Bromid kann mit den anderen Halogeniden nachgewiesen werden.
Nachweis von Bromid mit Schwefelsäure
[Bearbeiten]Eine weitere Möglichkeit ist, die Substanz mit konzentrierter Schwefelsäure zu erhitzen, wobei braune Dämpfe aufsteigen (elementares Brom). Hält man ein Filterpapier, das man mit Fluorescein (z. B. von einem gelben Textmarker) versehen und angefeuchtet hat, über das Reagenzglas, färben die braunen Dämpfe das Papier an der entsprechenden Stelle braunrot.
Chlor
[Bearbeiten]Chlor fällt in anorganischen Salzen als Chlorid, Chlorit, Hypochlorit, Chlorat und Perchlorat an.
Chlorid
[Bearbeiten]Chloride können mit den anderen Halogeniden nachgewiesen werden.
Perchlorat
[Bearbeiten]Perchlorate können als Kaliumperchlorat nachgewiesen werden.
Durchführung
[Bearbeiten]Ein Teil der Urprobe wird mit einer Kaliumlösung versetzt und danach gut abgekühlt (Eiswasser). Ein feinkristalliner Niederschlag zeigt das Vorliegen von Perchlorat an.

Iod
[Bearbeiten]Iod fällt in anorganischen Salzen als Iodidanion I− und Iodatanion IO3− an.
Iodid
[Bearbeiten]Iodide in Lösung ergeben, erhitzt mit konzentrierter Schwefelsäure, violette Dämpfe von elementarem Iod. Sie können mit den anderen Halogeniden nachgewiesen werden. Wichtig ist ebenfalls die Unterscheidung von Iodid und Iodat.
Unterscheidung von Iodid und Iodat (nach Kaiser)
[Bearbeiten]Die Unterscheidung von Iodid und Iodat nach Kaiser dient zum selektiven Nachweis von Iodid (auch Bromid) und Iodat nebeneinander in Substanzproben und −gemischen. Die Methode ist als Adaption und Verallgemeinerung der Anwendbarkeit der Iodidnachweise, die in Lehrbüchern, wie zum Beispiel Jander/Blasius: „Lehrbuch der analytischen und präparativen anorganischen Chemie“, aufgeführt werden, zu sehen.
Durchführung
[Bearbeiten]Die Probe (bei unlöslichen Proben der Sodaauszug) wird in einem Reagenzglas langsam mit verdünnter Salpetersäure (c ≈ 3 mol/L) leicht angesäuert, bis die Gasentwicklung aufhört (es kann auch mit Indikatorpapier nachgeprüft werden) und dann mit Chloroform unterschichtet. Färbt sich das Chloroform dabei braun oder violett, so ist dieses abzutrennen und die Probe zwei weitere Male mit Chloroform auszuschütteln, bis eine Färbung beim Unterschichten ausbleibt.
Die so behandelte Probe wird mit 30%igem Wasserstoffperoxid versetzt und geschüttelt, gegebenenfalls unter leichtem Erwärmen. Tritt hierbei eine violette Färbung des Chloroforms auf, so war Iodid zugegen; eine Braunfärbung weist auf Bromid hin. Liegen beide Halogenide vor, treten die Färbungen nacheinander auf.
Die angesäuerte, mit Chloroform unterschichtete Probe (s.o.) wird mit Zinkstaub versetzt und leicht erwärmt, womit nascierender Wasserstoff erzeugt wird. Eventuell vorhandenes Iodat wird von diesem zum Iod reduziert und das Chloroform färbt sich violett. Man sollte hierbei die Gasentwicklung im Reagenzglas im Auge behalten, da es unter Umständen zur Entzündung des Wasserstoffs am Brenner kommen kann.
Erklärung
[Bearbeiten]

Iodid und Bromid werden hierbei von Wasserstoffperoxid im sauren Millieu zu Iod bzw. Brom oxidiert, die jeweils die Färbungen des Chloroforms verursachen.

Fluorid
[Bearbeiten]Fluorid ist ein Halogenid, lässt sich jedoch nicht mit den gleichen Nachweisen wie die übrigen Halogenide aufspüren.
Bleitiegelprobe / Wassertropfenprobe
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Verdrängungsreaktion |
| pH | <7 schwefelsauer |
| Indikation | weißes Silicumoxid |
Durchführung
[Bearbeiten]
Zum Nachweis der Fluorid-Anionen wird eine kleine Portion der Probe in einen Bleitiegel gegeben, mit gepulverter Kieselsäure oder Natriumsilicat versetzt und vermischt. Anschließend überschichtet man vorsichtig mit Schwefelsäure. Es bildet sich das gasförmige Siliciumtetrafluorid.
Man verschließt den Tiegel mit einer PVC-Platte, an deren Unterseite sich ein kleiner Wassertropfen befindet (dieser darf natürlich nicht in die Probe hängen) und lässt ihn ungefähr eine Minute stehen. Das SiF4 reagiert mit dem Wasser wieder zu weißem Siliciumoxid, das sich kraterförmig im Tropfen absetzt.
Eine Alternative zur PVC-Platte mit Wassertropfen ist ein schwarzes Filterpapier, das angefeuchtet wird. Das entweichende SiF4-Gas zersetzt sich dort zu SiO2, was an einem weißen Fleck erkennbar ist.
Ohne Kieselgel oder Natriumsilicat kommt man bei der Bleitiegelprobe / Wassertropfenprobe aus, wenn man einen Deckel mit Öffnung verwendet, auf dem sich Glas befindet (z. B. Objektträger). Man befeuchtet die Probe im Bleitiegel mit Schwefelsäure, der entweichende Fluorwasserstoff ätzt das Glas an (vgl #Ätzprobe / Kriechprobe).
Erklärung
[Bearbeiten]- Fluorid-Ionen reagieren mit Schwefelsäure zu Sulfat-Ionen und Fluorwasserstoff.
- Siliciumdioxid reagiert mit Fluorwasserstoff zu Silicumtetrafluorid und Wasser.


Ätzprobe / Kriechprobe
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Verdrängungsreaktion |
| pH | <7 schwefelsauer |
| Indikation | angeätztes Glas |
Durchführung
[Bearbeiten]Für den Nachweis durch die Ätzprobe werden 10 bis 20 Tropfen konzentrierte Schwefelsäure in ein sauberes trockenes Reagenzglas gegeben. Um sich davon zu überzeugen, dass das Glas zu diesem Zeitpunkt noch nicht angeätzt ist, schüttelt man kurz. Dabei beobachtet man, dass die Schwefelsäure sich an den glatten Reagenzglaswänden wie ein viskoses Öl verhält. Anschließend gibt man eine fluoridhaltige Substanz zu. Man erhitzt nun für etwa fünf Minuten vorsichtig über der entleuchteten Bunsenbrennerflamme. Man stellt beim erneuten Umschwenken fest, dass sich ein deutlicher Abperl-Effekt an den nun aufgerauten Reagenzglaswänden zeigt. Das Reagenzglas ist hinterher nicht mehr brauchbar.
Erklärung
[Bearbeiten]- Fluorid-Ionen reagieren mit Schwefelsäure zu Sulfat-Ionen und Fluorwasserstoff.
Kalium
[Bearbeiten]Kalium verbleibt im Trennungsgang in der löslichen Gruppe. Es lässt sich wie die meisten Alkalikationen bevorzugt mittels Flammenfärbung aufspüren.
Nachweis als Kaliumperchlorat
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Fällungsreaktion |
| pH | egal |
| Indikation | weißer Niederschlag |
Durchführung
[Bearbeiten]Zur Probelösung werden einige Tropfen Perchlorsäure (65 %) gegeben. Es fällt ein weißer Niederschlag aus. Zur Sicherheit wird mit dem Niederschlag noch eine Flammenfärbung durchgeführt (Cobaltglas!). Falls ein Spektrometer zur Verfügung steht, wird natürlich auch dieses verwendet. Der Nachweis ist nicht sehr empfindlich.
Erklärung
[Bearbeiten]- Kalium-Ionen und Perchlorat-Ionen reagieren zum weißen Kaliumperchlorat.

Perchlorsäure ist die stärkste anorganische Säure und verdrängt alle anderen Säuren aus ihren Salzen.
Kupfer
[Bearbeiten]sulfide_neerslag.png)
Tipp: Kupfer(II)-salze färben Lösungen meist bläulich.
Eine mögliche Vorprobe ist die Borax und Phosphorsalzperle, welche in der Oxidationsflamme grün und in der Reduktionsflamme rötlich gefärbt ist.
Im Kationentrennungsgang fällt es in der Schwefelwasserstoffgruppe aus:
- Kupfer(II)-Kationen reagieren mit Schwefelwasserstoff zu schwarzem Kupfer(II)-sulfid

Nachweis als Kupfertetramminkomplex
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Komplexbildungsreaktion |
| pH | >>7 stark ammoniakalisch |
| Indikation | tiefblaue Lösung |
Durchführung
[Bearbeiten]Versetzt man die Analysenlösung mit Ammoniakwasser so bilden sich bei pH-Werten über 8 tiefblaue Komplexsalz-Lösungen (siehe Bild mittiges Reagenzglas).
Erklärung
[Bearbeiten]- Kupfer(II)-Ionen und Ammoniakwasser reagieren zum tiefblauen Komplex-Ion Tetraamminkupfer(II)
![{\displaystyle {\ce {Cu^{2+}(aq) \ + 4 NH_{4}^{+}(aq) \ + 4 OH^{-}(aq) -> [Cu(NH_3)_4]^{2+}\ + 4 H2O (l)}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/77f270db11c07aaa2d56ad82bf4f53f7d1f5b231)
Störungen
[Bearbeiten]Ni(II)-Ionen bilden ebenfalls blaue Komplexe, die nur geringfügig heller sind.
Nachweis als Kupferhexacyanidoferrat
[Bearbeiten]-Ionen1.jpg)
| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Komplexbildungsreaktion |
| pH | ? |
| Indikation | braunroter Niederschlag |
Durchführung
[Bearbeiten]Eine weitere Variante Kupfer(II)-Ionen nachzuweisen erfolgt mit einer Kaliumhexacyanidoferrat(II)-lösung (Gelbes Blutlaugensalz, früher: Kaliumhexacyanoferrat-II). Nach Zugabe von Blutlaugensalz zur Analysenlösung fällt ein braunroter Niederschlag aus (siehe Bild, rechtes Reagenzglas).
Erklärung
[Bearbeiten]- Kupfer(II)-Ionen und Kaliumhexacyanidoferrat(II) reagieren zum roten Komplex Kupfer(II)-hexacyanidoferrat(II) und Kalium-Ionen
![{\displaystyle \mathrm {2\ Cu^{2+}+K_{4}[Fe(CN)_{6}]\longrightarrow Cu_{2}[Fe(CN)_{6}]+4\ K^{+}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/6646fe3cf83d00107da7e86390d4fbb1e0ecc7ea)
Nachweis als Kupferthiocyanat
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Redoxreaktion, Komplexbildungsreaktion |
| pH | |
| Indikation | zunächst grün, im Überschuss schwarz, mit Sulfit weiß |
-Ionen%2Bthiocyanat.jpg)
Durchführung
[Bearbeiten]Bei Reaktion von Thiocyanat-Ionen mit Kupfer(II)-Ionen beobachtet man zunächst eine grüne Färbung der Lösung (siehe Bild linkes Reagenzglas). Beim Vorhandensein von Thiocyanat-Ionen oder Kupfer(II)-Ionen im Überschuss bildet sich ein schwarzer Niederschlag. (siehe Bild mittiges Reagenzglas) Wird der Niederschlag mit Sulfit-Ionen behandelt, so löst sich der schwarze Niederschlag und es bildet sich ein weißer Niederschlag. (Redoxreaktion, siehe Bild rechtes Reagenzglas)
Erklärung
[Bearbeiten]
Reaktion: Thiocyanat-Ionen reagieren mit Kupfer(II)-Ionen zu schwarzem, wasserunlöslichem Kupfer(II)-thiocyanat.

Reaktion: Schwarzes Kupfer(II)-thiocyanat reagiert mit Sulfit-Ionen im wässrigen Milieu zu weißem Kupfer(I)-thiocyanat, Thiocynanat-Ionen, Sulfat-Ionen und Wasserstoff-Ionen.

Lithium
[Bearbeiten]Lithium bleibt im Kationentrennungsgang in der löslichen Gruppe zurück. Wer versuchen möchte es zu fällen, kann versuchen, dies durch Eindampfen und Aufnahme mit konz. Salzsäure als Lithiumchlorid zu erreichen. Zum Abtrennen kann man sich zu nutze machen, dass es sich als einziges Alkalichlorid in Amylalkohol löst.
Nasschemische Nachweise von Lithium sind schwierig. Die karminrote Flammenfärbung ist der einzig wirklich einfache und sichere Nachweis, im Handspektroskop weist Lithium eine rote Linie bei 670,8 nm und eine schwächere orange bei 610,3 nm auf.
Nachweis als Lithiumphosphat
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Fällungsreaktion |
| pH | >>7 |
| Indikation | weißer Niederschlag |
Durchführung
[Bearbeiten]Die Lösung wird stark alkalisch gemacht und festes Dinatriumhydrogenphosphat hinzugegeben. Nach einiger Zeit sollte sich ein Niederschlag bilden, welcher im Sauren löslich ist. Ansonsten kann man versuchen, mehr Na2HPO4 hinzuzugeben. Die Lösung muss jedoch wirklich viel Li+ enthalten, damit es funktioniert.
Erklärung
[Bearbeiten]
Magnesium
[Bearbeiten]_reactions.JPG)
Magnesium verbleibt im Trennungsgang in der löslichen Gruppe. Erstes Anzeichen für Magnesium in der Probe kann eine sprühende Flamme (vgl. Wunderkerze) sein. Zum Nachweis muss stets sehr sauber abgetrennt werden, was Magnesium in Gemischen schwer nachweisbar macht.
Nachweis als Ammoniummagnesiumphosphat
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Fällungsreaktion |
| pH | >>7 NH4OH |
| Indikation | weißer Niederschlag |
Durchführung
[Bearbeiten]Die Lösung wird mit konz. Ammoniakwasser alkalisch gemacht und Ammoniumchlorid und Natriumhydrogenphosphat hinzuzugeben. Nach Erwärmen (~5 min) entsteht ein weißer Niederschlag. Unter dem Mikroskop sehen die Kristalle wie "Sargdeckel" aus: Vergleichsbild.
Erklärung
[Bearbeiten]

Störung
[Bearbeiten]Calcium, Strontium, Barium und Zink stören, weil sie ebenfalls einen Niederschlag bilden. Die Kristallform ist jedoch charakteristisch.
Nachweis als Chinalizarin-Farblack
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Farblack-Bildung |
| pH | <7 salzsauer |
| Indikation | Blaufärbung |
Durchführung
[Bearbeiten]Die mit verdünnter Salzsäure angesäuerte Lösung wird mit Chinalizarin-Lösung versetzt und mit Natronlauge stark alkalisiert. Falls ein kornblumenblauer Niederschlag ausfällt, ist dies ein positiver Hinweis auf Magnesium.
Störung
[Bearbeiten]Aluminium, Beryllium, Bor, Calcium, Cobalt, Fluoride, Gallium, Indium, Nickel, Zink stören.
Nachweis als Thiazolgelb-Farblack
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Farblack-Bildung |
| pH | <7 salzsauer |
| Indikation | Rotfärbung |
Durchführung
[Bearbeiten]Die Probe wird in Wasser gelöst und mit verdünnte Salzsäure angesäuert. Anschließend wird sie mit einem Tropfen der Thiazolgelb-Lösung (auch Titangelb genannt, obwohl kein Titan vorkommt) versetzt und mit verdünnter Natronlauge alkalisch gemacht. Bei Anwesenheit von Magnesium entsteht ein hellroter Niederschlag.
Störung
[Bearbeiten]Nickel-, Zink-, Mangan- und Cobalt-Ionen stören diesen Nachweis und sollten vorher als Sulfide ausgefällt werden.
Mangan
[Bearbeiten]Nachweis als Braunstein
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Redoxreaktion |
| pH | >>7 |
| Indikation | schwarz-braune Lösung |
Durchführung
[Bearbeiten]Mangan(II)-Kationen werden zum Nachweis im Kationentrenngang im so genannten „Alkalischen Bad“ – einer Mischung aus konz. Wasserstoffperoxid und Natronlauge – zum Mangan(IV)-Kation oxidiert.
Erklärung
[Bearbeiten]- Farblose Mangan(II)-Ionen reagieren mit Wasserstoffperoxid in alkalischer Lösung zu braunem Manganoxidhydroxid (Braunstein) und Wasser.

Nachweis als Permanganat
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Redoxreaktion |
| pH | <7 |
| Indikation | violette Lösung |
Das Mangan(IV)-oxid-hydroxid „Braunstein“ wird dann durch Kochen in konz. Salpetersäure gelöst und mit Blei(IV)-oxid zum violetten Permanganat aufoxidiert:
- Braune Mangan(IV)-Ionen reagieren mit Blei(IV)-oxid in Wasser zu violetten Permanganat-Ionen, Blei(II)-Ionen und Wasserstoff-Ionen.

Ist die Aufschwemmung durch Blei(IV)-oxid zu dunkel, füllt man vorsichtig etwas Wasser auf, welches sich verfärbt. Das Verfahren eignet sich auch als Vorprobe, wird aber von Iodid gestört.
Nachweis über Oxidationsschmelze
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Redoxreaktion |
| pH | entfällt |
| Indikation | blaugrüne Salze |
Durchführung
[Bearbeiten]Nachweis über die Oxidationsschmelze: Die Probe wird mit einer stöchiometrisch angepassten Menge eines Soda-Salpeter-Salzgemisches gemörsert und im Porzellantiegel erhitzt - blaugrünes Manganat(VI) zeigt Mangansalze an.
Tipp: Das Manganat(VI) disproportioniert bei Kontakt mit Essigsäure zum rosavioletten Permanganat und braunen Mangan(IV)-oxid (Redoxreaktion).
Erklärung
[Bearbeiten]- Mangan(IV)oxid reagiert mit Natriumcarbonat und Kaliumnitrat zu Natriummanganat(VI) (blaugrün), Kohlenstoffdioxid und Kaliumnitrit.

Molybdän
[Bearbeiten]Molybdän kommt im KTG in der Schwefelwasserstoffgruppe, genauer in der Arsen-Gruppe vor.
Vorprobe als Molybdänblau
[Bearbeiten]Man kocht etwas Ursubstanz mit wenig Zinnchlorid und 20 mL konz. Schwefelsäure in einer offenen Schale fast bis zur Trockene ab. Beim Erkalten tritt eine intensive Blaufärbung ein, die von einem Oxid der ungefähren Zusammensetzung Mo3O8 (=MoO3·Mo2O5) hervorgerufen wird.
Störungen
[Bearbeiten]Wolfram bildet ein himmelblaues Oxid (Wolframblau) und Vanadium zeigt ebenfalls eine hellblaue Färbung.
Nachweis als Molybdophosphat
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Fällungsreaktion |
| pH | <<7 salpetersauer |
| Indikation | gelbe Kristalle |
Durchführung
[Bearbeiten]Die stark salpetersaure Lösung wird in einem kleinen Reagenzglas mit wenig Ammoniumchlorid bzw. Kaliumchlorid sowie 1-2 Tropfen 2 mol/L Natriumhydrogenphosphat versetzt und erwärmt. Es scheiden sich äußerst feine gelbe Kristalle von Ammonium- bzw. Kaliummolybdophosphat ab.
Erklärung
[Bearbeiten]- Molybdän-Ionen reagieren mit Natriumhydrogenphosphat und Ammoniumchlorid zu gelbem Ammoniummolybdophosphat, welches ausfällt, sowie Natriumchlorid und Salzsäure.
![{\displaystyle {\begin{array}{ll}&\mathrm {Mo^{6+}+Na_{2}HPO_{4}+3\ NH_{4}Cl+H_{2}O} \\\longrightarrow &\mathrm {(NH_{4})_{3}[Mo(PO_{4})]\downarrow +2\ NaCl+Cl^{-}+H_{3}O^{+}} \end{array}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5ab924908dc72c8ea982172fc89a0e739b989510)
- Molybdän-Ionen reagieren mit Natriumhydrogenphosphat und Kaliumchlorid zu gelbem Kaliummolybdophosphat, welches ausfällt, sowie Natriumchlorid und Salzsäure.
![{\displaystyle {\begin{array}{ll}&\mathrm {Mo^{6+}+Na_{2}HPO_{4}+3\ KCl+H_{2}O} \\\longrightarrow &\mathrm {K_{3}[Mo(PO_{4})]\downarrow +2\ NaCl+Cl^{-}+H_{3}O^{+}} \end{array}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/833a84230dac6517aa9da9910f40fa90e358b926)
Natrium
[Bearbeiten]Natrium verbleibt im Trennungsgang in der löslichen Gruppe. Es lässt sich wie die meisten Alkalikationen bevorzugt mittels Flammenfärbung aufspüren. Dort muss es jedoch lange (mindestens 3 Minuten) zu sehen sein, da Natriumverunreinigungen überall vorkommen (z.B. Handschweiß). Weil fast alle Natriumsalze gut löslich sind, ist es schwierig Natrium nasschemisch mit Fällungsreaktionen zu finden.
Nachweis als Natriumhexahydroxoantimonat(V)
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Fällungsreaktion |
| pH | >>7 KOH |
| Indikation | weißer feinkristalliner Niederschlag |
Durchführung
[Bearbeiten]Die Lösung wird stark alkalisch gemacht und auf einem Uhrglas mit einigen Tropfen K[Sb(OH)6]-Lösung versetzt. Nach einer Viertelstunde bildet sich ein weißer feinkristalliner Niederschlag, der sich mit Wasser nicht abspülen lässt und sich sandig anfühlt.
Erklärung
[Bearbeiten]![{\displaystyle \mathrm {Na^{+}+[Sb(OH)_{6}]^{-}\longrightarrow Na[Sb(OH)_{6}]\downarrow } }](https://wikimedia.org/api/rest_v1/media/math/render/svg/da5ce9b2973150c8270fea63dfc19999c0f4fafc)
Störung
[Bearbeiten]Lithium und Ammonium stören, weil sie ebenfalls einen Niederschlag bilden. Der Versuch funktioniert nur mit konzentrierter Natriumlösung
Nachweis als Natrium-Magnesium-triuranyl-nonaacetat
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Fällungsreaktion |
| pH | <7 essigsauer |
| Indikation | gelbe Kristalle |
Durchführung
[Bearbeiten]Die Lösung wird essigsauer gemacht und mit ein paar Tropfen Magnesiumuranylacetat-Lösung versetzt. Nach einiger Zeit fallen gelbe Kristalle aus.
Erklärung
[Bearbeiten]
Störung
[Bearbeiten]Lithium stört, weil es ebenfalls einen Niederschlag bildet.
Nickel
[Bearbeiten]_reactions.JPG)
(2) schwarzes Nickelsulfid,
(3) hellgrünes Nickelhydroxid,
(4) blaues Hexaaminnickel,
(5) basische grüne Carbonatsalze wechselnder Zusammensetzung
Nickelsalze fallen im Trennungsgang in der Ammoniumsulfidgruppe als schwarze Sulfide aus.
- Nickel(II)-Ionen reagieren mit Ammoniumsulfid zu Nickel(II)sulfid und Ammonium-Ionen.

Des Weiteren ist die Fällung des Hydroxids möglich, das man an seiner spezifisch grünen Farbe erkennen kann. Als Fällungsmittel wird meist Natronlauge verwendet.
- Nickel(II)-Ionen reagieren mit Natronlauge zu grünem, wasserunlöslichem Nickel(II)hydroxid und Natrium-Ionen.

Durch Zugabe von starken Oxidationsmitteln wie Chlor oder Brom, jedoch nicht mit Wasserstoffperoxid, geht das grüne Hydroxid in ein höheres, schwarzes Oxid über.
- Grünes Nickel(II)hydroxid reagiert mit Brom zu schwarzem, wasserunlöslichem Nickel(IV)oxid und Bromwasserstoff.

Wird zu einer Probelösung, die Nickel(II)-Ionen enthalten soll, Ammoniak-Lösung zugetropft, kann man beobachten, das sich zuerst ein grüner Niederschlag von Nickel(II)-hydroxid bildet, der bei Überschuss von Ammoniak sich unter Blaufärbung wieder auflöst.
- Nickel(II)-Ionen reagieren im wässrigen Milieu mit Ammoniakwasser zu grünem, wasserunlöslichem Nickel(II)hydroxid und Ammonium-Ionen.

- Nickel(II)-Ionen reagieren bei Überschuss von Ammoniakwasser zu blauen, wasserlöslichen Hexaaminnickel(II)-Ionen und Hydroxid-Ionen.
![{\displaystyle \mathrm {Ni(OH)_{2}+6\ NH_{3}\longrightarrow [Ni(NH_{3})_{6}]^{2+}+2\ OH^{-}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/8d3dc3dab60788331ef83ba195b23690087e644d)
Nachweis mit DAD
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Chelatkomplexbildung |
| pH | >7 alkalisch |
| Indikation | himbeerroter Niederschlag |
Durchführung
[Bearbeiten]Man versetzt die zuvor alkalische gemachte Analysenlösung mit einer alkoholischen Lösung von Diacetyldioxim (=Dimethylglyoxim), die auch Tschugajews Reagens genannt wird. Es bildet sich ein himbeerroter voluminöser Niederschlag, der in verdünnten Mineralsäuren wieder zerfällt. In Natronlauge und in Gegenwart von starken Oxidationsmitteln wie Peroxodisulfat erscheint eine ebenfalls intensiv rote, jedoch lösliche Nickel(III)-Verbindung.
Erklärung
[Bearbeiten]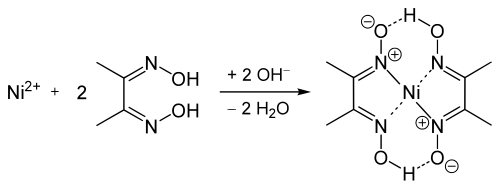
- Diacetyldioxim bildet mit Nickel in alkalischer Lösung einen Chelatkomplex.

Stickstoff
[Bearbeiten]Anorganisch gebundener Stickstoff kommt als NH4+ Ammoniumkation sowie in den Anionen Cyanid CN−, Thiocyanat SCN−, Nitrat NO3– und Nitrit NO2– vor.
Ammonium
[Bearbeiten]Nachweis mittels Kreuzprobe
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Verdrängungsreaktion |
| pH | >7 alkalisch |
| Indikation | verfärbtes Indikatorpapier |

Durchführung
[Bearbeiten]In einem Uhrglas wird angefeuchtetes Universalindikatorpapier befestigt, in ein zweites Uhrglas gibt man die Probe, etwas Natronlauge und einige Tropfen Wasser und bedeckt nun das zweite Uhrglas mit dem ersten. Eine Blaufärbung des Indikatorpapiers zeigt Hydroxidionen, die durch Ammoniak entstanden sind, das aus dem Ammoniumsalz freigesetzt wurde. Das Indikatorpapier kann dabei nicht die Lauge, die durch das NaOH eingebracht wurde, anzeigen, weil dieses die wässrige Lösung nicht verlassen können. Das Indikatorpapier sollte also nicht in die Lösung fallen. Zum Vergleich legt man meist außerhalb der beiden Uhrgläser nochmal einen Streifen Indikatorpapier darüber und befeuchtet ihn mit dest. Wasser, das auch schon leicht alkalisch ist.
Erklärung
[Bearbeiten]- Ammonium-Ionen und Hydroxid-Ionen reagieren zu gasförmigem Ammoniak und Wasser.

Nachweis mittels Neßlers-Reagenz
[Bearbeiten]Durchführung
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Komplexbildung |
| pH | >7 alkalisch |
| Indikation | gelbbrauner Niederschlag |
Beim Nachweis mit der Neßlers-Reagenz wird eine Kaliumtetraiodomercurat(II)-lösung mit Natronlauge alkalisiert. Die Probelösung, die auf Ammonium-Ionen untersucht werden soll, wird mit wenig Neßlers Reagenz umgesetzt. Bei gelbbrauner Färbung bzw. brauner Ausflockung sind Ammonium-Ionen nachgewiesen, es entsteht das Iodidsalz der Millonsche Base.
Erklärung
[Bearbeiten]- Ammonium-Ionen, Kaliumtetraiodomercurat(II), Natronlauge und Hydroxid-Ionen reagieren zum Iodidsalz der Millonschen Base, die in wässriger Lösung ausflockt, Kaliumiodid, Natriumiodid und Wasser.
![{\displaystyle {\begin{array}{ll}&\mathrm {NH_{4}^{+}+2\ K_{2}[HgI_{4}]+3\ NaOH+OH^{-}} \\\longrightarrow &\mathrm {[Hg_{2}N]I\downarrow +4\ KI+3\ NaI+4\ H_{2}O} \end{array}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/7322a01d47f3d90319435397ff8256827da8356c)
Cyanid
[Bearbeiten]Nachweis als Berliner Blau
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Komplexbildung |
| pH | 8-9 |
| Indikation | tiefblaue Lösung |
Durchführung
[Bearbeiten]Zur Prüfung auf Cyanid-Ionen in einer Probe wird, sofern noch nicht geschehen, die Probe mit Natronlauge auf pH-Wert 8 bis 9 alkalisiert. Anschließend wird Eisen(II)-sulfatlösung im Unterschuss hinzugegeben und diese Mischung mit fächelnder Flamme bis zur Trocknung eingedampft (Arbeit unter dem Abzug unabdingbar! Giftige Cyaniddämpfe können entweichen!). Anschließend wird der Rückstand mit verdünnter Salzsäure gelöst, es entsteht eine klare Lösung, die mit verdünnter Eisen(III)chloridlösung versetzt wird. Bei Anwesenheit von Cyanid bildet sich das tiefblaue Pigment Berliner Blau.
Erklärung
[Bearbeiten]- Cyanid-Ionen reagieren mit Eisen(II)-Ionen zu Hexacyanidoferrat(II)-Ionen.
![{\displaystyle \mathrm {6\ CN^{-}+Fe^{2+}\longrightarrow [Fe(CN)_{6}]^{4-}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/606c5a981c6b1fa7a83c7e8542a83cd9ec0e3dfe)
- Hexacyanoferrat(II)-Ionen reagieren mit Eisen(III)-Ionen zu blauem Eisen(III)hexacyanoferrat(II).
![{\displaystyle \mathrm {3\ [Fe(CN)_{6}]^{4-}+4\ Fe^{3+}\longrightarrow Fe_{4}[Fe(CN)_{6}]_{3}\downarrow } }](https://wikimedia.org/api/rest_v1/media/math/render/svg/6eb876ffa35fc2f72db27208ce32e78a08e1ced4)
Nachweis mit Polysulfiden
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Komplexbildung |
| pH | |
| Indikation | tiefrote Lösung |
Durchführung
[Bearbeiten]Alternativ können Cyanide mit Polysulfiden zu Thiocyanat umgesetzt werden. Dazu werden einige Tropfen Ammoniumpolysulfidlösung (gelbes Ammoniumsulfid) zu der in Wasser gelösten Ursubstanz gegeben. Die Lösung wird bis zur Trocknung erhitzt und der Rückstand in wenig verdünnter Salzsäure suspendiert. Anschließend wird filtriert. Die klare Lösung wird mit wenig verdünnter Eisen(III)-chloridlösung versetzt. Beim Entstehen einer tiefroten Färbung, hervorgerufen durch Eisen(III)-thiocyanat, war Cyanid zugegen.
Erklärung
[Bearbeiten]- Cyanid-Ionen reagieren mit Polysulfid-Ionen zu Thiocyanat-Ionen und Sulfid-Ionen.

- Thiocyanat-Ionen reagieren mit Eisen(III)-Ionen zu blutrotem Eisen(III)-thiocyanat.

Thiocyanat
[Bearbeiten]Stierblutprobe
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Komplexbildung |
| pH | |
| Indikation | tiefrote Färbung |
Thiocyanat-, oder auch Rhodanid-Ionen werden qualitativ mit der „Stierblutprobe“ nachgewiesen. Diese Reaktion wird auch zum Nachweis von Eisen mit Thiocyanatlösung eingesetzt.
Durchführung
[Bearbeiten]Dabei wird der zu untersuchenden Lösung eine gesättigte Eisen(III)-chlorid-Lösung zugegeben. Erscheint eine intensiv „stierblutrote“ Färbung, so waren Thiocyanat-Ionen vorhanden.
Erklärung
[Bearbeiten]- Reaktion: Thiocyanat-Ionen und Eisen(III)-Ionen reagieren im wässrigen Milieu zum Komplex Pentaaquathiocyanatoferrat(III), welcher blutrot erscheint.
![{\displaystyle \mathrm {SCN^{-}+Fe^{3+}+5\ H_{2}O\longrightarrow [Fe(SCN)(H_{2}O)_{5}]_{aq}^{2+}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/040e33dec80c808c7b4d5d5c5dbd5f282bc16b93)
Nachweis mit Kupfersulfat
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Redoxreaktion, Komplexbildung |
| pH | |
| Indikation | zunächst grün, im Überschuss schwarz, mit Sulfit weiß |
Ein weiterer spezifischer Nachweis kann mit Kupfersulfatlösung erfolgen.
Durchführung
[Bearbeiten]Zur in Wasser gelösten Ursubstanz wird frisch bereitete Kupfer(II)-sulfat zugegeben. Bei Reaktion von Thiocyanat-Ionen mit Kupfer(II)-Ionen beobachtet man zunächst eine grüne Färbung der Lösung (siehe Bild linkes Reagenzglas). Beim Vorhandensein von Thiocyanat-Ionen oder Kupfer(II)-Ionen im Überschuss bildet sich ein schwarzer Niederschlag. (siehe Bild mittiges Reagenzglas) Wird der Niederschlag mit Sulfit-Ionen behandelt, so löst sich der schwarze Niederschlag und es bildet sich ein weißer NS. (Redoxreaktion, siehe Bild rechtes Reagenzglas)
Erklärung
[Bearbeiten]Reaktion: Thiocyanat-Ionen reagieren mit Kupfer(II)-Ionen zu schwarzem, wasserunlöslichem Kupfer(II)-thiocyanat.
Reaktion: Schwarzes Kupfer(II)-thiocyanat reagiert mit Sulfit-Ionen im wässrigen Milieu zu weißem Kupfer(I)-thiocyanat, Thiocyanat-Ionen, Sulfat-Ionen und Oxonium-Ionen.

Nitrat
[Bearbeiten]Nitratnachweis mittels Ringprobe
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Redoxreaktion, Komplexbildung |
| pH | <<7 schwefelsauer |
| Indikation | brauner Ring |

Durchführung
[Bearbeiten]Zum Nitratnachweis mittels Ringprobe wird die Analysensubstanz mit einigen Tropfen Eisen(II)-sulfat-Lösung und verdünnter Schwefelsäure versetzt. Anschließend hält man das Reagenzglas schräg und lässt am Rand vorsichtig einige Tropfen konzentrierter Schwefelsäure herunterlaufen, um die Lösung zu unterschichten. Eine ringförmige Braunfärbung an der Grenzschicht zeigt Nitrat an. Zum besseren Erkennen bei geringer Konzentration hält man das Reagenzglas vor einen weißen Kittelärmel oder gegen ein Blatt Papier. Je nach Konzentration kann der Ring auch bis auf ein fahles Violett verringert sein, es ist daher eine Negativprobe zum Vergleich sehr hilfreich.
Erklärung
[Bearbeiten]An der Schichtgrenze von Probelösung und Schwefelsäure findet eine Redoxreaktion statt:
- Nitrat-Ionen werden zu Stickstoffmonoxid reduziert und die Eisen(II)-Ionen zu Eisen(III)-Ionen oxidiert

Im weiteren Reaktionsverlauf bildet sich ein Komplex, der für die Braunfärbung sorgt, die namensgebend für die Nachweisreaktion ist:
- Aus Eisen(II)-Ionen an den sich Stickstoffmonoxid angelagert hat, bildet sich in wässriger Lösung der Pentaaquanitrosyleisen(II)-Komplex
![{\displaystyle \mathrm {[Fe(H_{2}O)_{6}]^{2+}+NO\rightarrow [Fe(H_{2}O)_{5}NO]^{2+}+H_{2}O} \!}](https://wikimedia.org/api/rest_v1/media/math/render/svg/478a589cd86c93f8822c7d9fc8ad73a71133173f)
Störung
[Bearbeiten]Es kann zu Konzentrationsniederschlag entlang der Schichtgrenze kommen. Diese ist meist weiß und lässt subtil positive Ergebnisse nicht mehr erkennen.
Nitrit-Ionen stören ebenfalls diesen Nachweis, da sich die Lösung bereits bei der Zugabe der Eisen(II)-Lösung braun färbt. Durch Kochen mit Harnstoff werden anwesende Nitrit-Ionen beseitigt.
- Salpetrige Säure und Harnstoff reagieren zu Stickstoff, Kohlendioxid und Wasser

Nitratnachweise mit Lunges Reagenzien
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Redoxreaktionen, Bildung eines Azofarbstoffes |
| pH | < essigsauer |
| Indikation | rote Farbe |
Durchführung
[Bearbeiten]Bei dieser Reaktion muss die Lösung Nitritionen-frei sein. Man kann entweder eine Abtrennung (siehe #Nitratnachweis mittels Ringprobe) probieren oder vor der Zugabe von Zink die Nitritionen mit Amidoschwefelsäure zu Stickstoff reduzieren.
Die Lösung wird, wenn sie sauer ist, mit Carbonationen neutralisiert und anschließend mit Essigsäure, auf einer Tüpfelplatte, angesäuert. Danach kommen einige Tropfen Sulfanilsäure und ein Kristall 1-Naphthylamin hinzu. Es darf an dieser Stelle keine Färbung auftreten, ansonsten ist die Probelösung nitrithaltig, was mit Zugabe von Harnstoff behoben werden muss. Nun wird noch etwas Zinkstaub hinzugegeben, der Nitrationen zu Nitritionen reduziert und für eine langsame gelb-orange Färbung der Lösung und eine rote Färbung des Kristalls sorgt.
Erklärung
[Bearbeiten]- Nitrat wird durch Zinkstaub und Eisessig (Ethansäure) zu Nitrit reduziert.

- Salpetrige Säure wird Amidoschwefelsäure zu Schwefelsäure, Stickstoff und Wasser reduziert

Der Rest ist analog zum Nitritnachweis mit Lunges Reagenzien.
Störung
[Bearbeiten]Der Nachweis wird durch die Anwesenheit von Nitrit-, Sulfit-, Thiosulfat- und Hexacyanoferrat(III)-Ionen gestört.
Nitrit
[Bearbeiten]Nitrit (NO2-) kommt als Salz der Salpetrigen Säure (HNO2) vor.
Nitritnachweis mit Lunges Reagenzien
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Bildunges eines Azofarbstoffes |
| pH | <7 schwach sauer |
| Indikation | Rotfärbung |
Durchführung
[Bearbeiten]Die Probelösung muss bei Untersuchung auf Nitrit-Ionen keine besonderen Eigenschaften besitzen. Sie sollte nur nicht zu sauer sein, ansonsten muss die Lösung mit Carbonat-Ionen neutralisiert werden. Nun wird die Probelösung wiederum mit stark konzentrierter Essigsäure angesäuert. Danach kommen jeweils 2–3 Tropfen Sulfanilsäure (Lunge I) hinzu und ein Kristall 1-Naphthylamin (Lunge II). Nimmt der Kristall an dieser Stelle eine rote Färbung an, so sind Nitrit-Ionen in der Lösung enthalten.
Erklärung
[Bearbeiten]Durch Zugabe von Sulfanilsäure (1) und 1-Naphthylamin (3) bildet sich zuerst ein Diazoniumsalz (2), das mit Naphthylamin weiter zu einem Azofarbstoff (4) reagiert und die Lösung sehr schnell rot färbt:

Permanganat
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Redoxreaktion, Iodprobe |
| pH | <<7 schwefelsauer |
| Indikation | Gelbfärbung, mit Stärke intensiv blau bis schwarz |
Um Permanganat-Ionen nachzuweisen, gibt es eine Reihe von Experimenten, wobei immer auf die Fähigkeit des MnO4−, Stoffe zu oxidieren, zurückgegriffen wird.
Nachweis mit Iodid
[Bearbeiten]Eine Reaktion, bei der man sehr schön erkennen kann, dass es sich um Permanganat-Ionen handelt, ist die Oxidation von Iodid-Ionen in einer Kaliumiodid-Lösung.
Durchführung
[Bearbeiten]Der Probelösung (schwach violett bis violett, schwefelsauer) wird eine gesättigte Kaliumiodidlösung zugegeben. Eine Gelbfärbung ist erstes Anzeichen für die Oxidation der Iodid-Ionen zu elementarem Iod. Um jedoch sicher zu sein, wird etwas Stärke zu der vermeintlichen Iod-Kaliumiodid-Lösung gegeben: Eine intensive blaue bis schwarze Färbung zeigt elementares Iod an.
Erklärung
[Bearbeiten]- Permangant-Ionen, Iodid-Ionen und Oxonium-Ionen reagieren zu Iod, Mangan(II)-Ionen und Wasser.

- Iod (gelb) und Stärke (weiß) reagieren zu Iodstärke (blau-schwarz), welches in wässriger Lösung ausfällt.
![{\displaystyle \mathrm {n\ I_{2}+-[C_{6}H_{10}O_{5}]_{n}-\longrightarrow -[C_{6}H_{10}O_{5}(I_{2})]_{n}-\downarrow } }](https://wikimedia.org/api/rest_v1/media/math/render/svg/49333ded1a1872afd85860117ce68af714a5dcb1)
Die quantitative und qualitative Bestimmung von Phosphor erfolgt über das Phosphat (genauer Orthophosphat PO43−). Gebundener Phosphor wird hierzu gegebenenfalls durch oxidierenden Aufschluss in Phosphat überführt.
Phosphat
[Bearbeiten]Phosphat als Zirkoniumphosphat
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Fällungsreaktion |
| pH | << 7 stark salzsauer |
| Indikation | durchsichtiger Niederschlag |
Durchführung
[Bearbeiten]Phosphat (PO43–) lässt sich als Zirkoniumphosphat nachweisen. Dazu wird die stark salzsaure Analysenlösung mit möglichst frisch hergestellter Lösung von Zirconiumoxidchlorid auch Zirkonylchlorid (ZrOCl2) oder Zirconiumoxidnitrat auch Zirkonylnitrat (ZrO(NO3)2) versetzt. Es fällt ein durchsichtig-milchiger, gallertartiger, flockiger Niederschlag aus. Bei verdünnten Lösungen kann ein Erwärmen der Probe die Reaktion beschleunigen.
Erklärung
[Bearbeiten]- Phosphat und Zirconylchlorid reagieren in saurer Lösung zu einem milchigen Niederschlag von Zirkoniumhydrogenphosphat, Wasser und Salzsäure.

Nachweis mit Molybdatlösung
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Komplexbildung |
| pH | <7 |
| Indikation | gelber Niederschlag |
Durchführung
[Bearbeiten]Die schwermetallfreie Probelösung wird mit konzentrierter Salpetersäure aufgekocht (Oxidation störender Reduktionsmittel), mit Ammoniummolybdatlösung versetzt und erneut kurz aufgekocht. Dabei weist eine gelbe Trübung auf Phosphate hin, die mit Molybdaten den gelben Ammoniummolybdatophosphatkomplex bilden.
Erklärung
[Bearbeiten]Unter Berücksichtigung, dass Ammoniumheptamolybdat in wässriger Lösung ein Gleichgewicht eingeht:

ergibt sich folgende Reaktionsgleichung:
- Ammoniummolybdat und Phosphationen bilden in sauer Lösung den gelben Ammoniummolybdatophosphatkomplex
![{\displaystyle \mathrm {\ PO_{4}^{3-}+12\ MoO_{4}^{2-}+24\ H_{3}O^{+}+3\ NH_{4}^{+}\longrightarrow (NH_{4})_{3}[P(Mo_{3}O_{10})_{4}]+36\ H_{2}O} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/27736f3c7169966ca34079afb20049c17f32c6c1)
Störung
[Bearbeiten]In Anwesenheit reduzierender Ionen wie Sulfid, Bromid, Iodid, Thiosulfat oder auch Zinn(II)-Kationen entsteht stattdessen Molybdänblau.
Nachweis mit ammoniakalischer Magnesiumsalzlösung
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Fällungsreaktion |
| pH | >7 |
| Indikation | weißer Niederschlag |
Durchführung
[Bearbeiten]Die schwermetallfreie, mit Ammoniak und Ammoniumchlorid auf pH 8–9 gepufferte Probelösung wird mit Magnesiumchloridlösung versetzt. Eine weiße Trübung von Magnesiumammoniumphosphat (MgNH4PO4 · 6 H2O) zeigt ebenfalls Phosphat an (säurelöslich):
Erklärung
[Bearbeiten]- Magnesiumssalze bilden in ammoniakalischer Lösung Magnesiumammoniumphosphat (weiß).
Quecksilber
[Bearbeiten]Quecksilber wird im Kationentrennungsgang in der Salzsäuregruppe abgetrennt und fällt gegebenenfalls auch in der Schwefelwasserstoffgruppe als schwarzes Sulfid aus.
Fast alle anorganischen Quecksilbersalze sind hochgiftig. Elementares Quecksilber verdampft bei Zimmertemperatur. Eingeatmete Dämpfe sind ebenfalls stark toxisch und führen zur Quecksilbervergiftung.
Nachweis mit Ammoniak
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Disproportionierung |
| pH | <7 salz- und salpetersauer |
| Indikation | schwarzer Filter |
Durchführung
[Bearbeiten]Löst man die Analysensubstanz in HNO3 und versetzt mit HCl liegt Quecksilber als Quecksilber(I)-chlorid (Hg2Cl2) vor (passiert im Kationentrennungsgang in der Salzsäuregruppe). Versetzt man nun mit halbkonzentriertem Ammoniak, so färbt sich der Filter schwarz.
Erklärung
[Bearbeiten]Es entsteht ein Gemisch von weißem Quecksilber(II)-amidochlorid ("unschmelzbares Präzipitat") und feinverteiltem, schwarzem Quecksilber, welches den Niederschlag schwarz färbt.
- Quecksilber(I)-chlorid und Ammoniak reagiert zu elementarem Quecksilber (schwarz) und Quecksilber(II)-amidochlorid (weiß) und Ammoniumchlorid

Amalgamprobe
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Redoxreaktion |
| pH | |
| Indikation | silbriger Belag |
Durchführung
[Bearbeiten]Ein Kupferblechschnipsel wird auf einem Uhrglas unter dem Abzug mit einem kleinen Tropfen der gelösten Analysensubstanz befeuchtet. Nach einigen Minuten sitzt auf dem Kupfer-Blech ein silbriger Belag, der beim Polieren mit einem Filterbausch silberglänzend wird (Bildung von Amalgam nach Redoxreaktion). Wenn der Belag vollständig abgerieben werden kann, liegt kein Quecksilber sondern ausschließlich Silber vor.
Erklärung
[Bearbeiten]- Quecksilberkationen oxidieren Kupfer zu Kupferionen und Quecksilber; dieses bildet mit weiterem Kupfer ein Amalgam aus..

Nachweis als Cobaltthiocyanatomercurat(II)
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Fällungsreaktion |
| pH | |
| Indikation | blaue Kristalle |
Durchführung
[Bearbeiten]Quecksilber(II)-Kationen können auch mit einer cobalthaltigen Thiocyanat-Lösung nachgewiesen werden. Dazu wird 1 Tropfen der Lösung auf dem Objektträger mit 1 Tropfen 14 mol/l Salpetersäure vorsichtig zur Trockne eingedampft. Der Rückstand wird mit 1 Tropfen 1 mol/l Essigsäure und danach mit einem kleinen Tropfen Reagenzlösung versetzt, wobei die Reagenzlösung aus 3,3 g Ammoniumthiocyanat und 3 g Cobaltnitrat welches zusammen in 5 ml Wasser gelöst wurde, besteht. Die Bildung blauer, keilförmiger Kristalle von Cobaltthiocyanatomercurat(II) zeigt Quecksilber(II)-Ionen an.
Erklärung
[Bearbeiten]- Quecksilber-Ionen, Cobalt-Ionen und Thiocyanat-Ionen reagieren zum blauen, keilförmigen Cobaltthiocyanatomercurat(II).
![{\displaystyle \mathrm {Hg^{2+}+Co^{2+}+4\ SCN^{-}\longrightarrow Co[Hg(SCN)_{4}]\downarrow } }](https://wikimedia.org/api/rest_v1/media/math/render/svg/9e62ceae0c28c55c25fc0c51713ab45df46806dc)
Silber
[Bearbeiten]Silber wird im Kationentrennungsgang in der Salzsäuregruppe abgetrennt. Durch Ansäuern der Diamminsilberchlorid-Lösung (z.B. mit HNO3) lässt es sich nachweisen, wobei farbloses Silberchlorid (AgCl) ausfällt.
Nachweis mit Chloridlösung
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Fällungsreaktion, |
| pH | <7 salpetersauer |
| Indikation | weißer Niederschlag, der im Überschuss oder durch Zugabe von Ammoniak verschwindet |
- Der Nachweis erfolgt analog zu den Halogenidnachweisen mit Silbersalzslösung

Durchführung
[Bearbeiten]Der Nachweis erfolgt durch Zugabe einer wässrigen Chloridlösung zur Stoffprobe. Chlorid-Ionen bilden mit Silber(I)-Ionen einen weißen, käsigen Niederschlag, der sehr lichtempfindlich ist und sich nach einiger Zeit infolge der Zersetzung von Silber(I)-chlorid in freies Chlor und feinverteiltem, kolloidalem Silber blaugrau verfärbt. Silber(I)-chlorid löst sich jedoch im Chlorid-Überschuss unter Bildung eines Dichloroargentat-Komplexes. Auch löst sich Silber(I)-chlorid in verdünntem Ammoniakwasser unter Bildung des Amminkomplexes wieder auf.
Erklärung
[Bearbeiten]- Silber(I)-Ionen und Chlorid-Ionen reagieren in wässriger Lösung zum einem weißen Niederschlag von Silber(I)-chlorid.

- Silberchlorid reagiert bei Chlorid-Ionen-Überschuss zum löslichen Dichloroargentat(I)-komplexion.
![{\displaystyle \mathrm {AgCl+Cl^{-}\longrightarrow [AgCl_{2}]^{-}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/79a6e84e10f23297774dbbe8fccb33bbfdbcaf3d)
- Das in Wasser unlösliche Silber(I)-chlorid reagiert nach Zugabe von Ammoniakwasser zu einem farblosen Komplex, dem Diamminsilber(I)-komplex und Chlorid-Ionen.
Nachweis mit Cyanid oder Thiocyanat
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Komplexbildung |
| pH | 7 neutral |
| Indikation | weißer Niederschlag, im Überschuss löslich |
Durchführung
[Bearbeiten]Eine weitere Methode ist die Fällung mit Cyanid-Ionen bzw. Thiocyanat-Ionen in neutraler Lösung. Sie sind nicht säurelöslich, lösen sich jedoch im Fällungsmittel unter Bildung der komplexen Anionen.
Erklärung
[Bearbeiten]- Silber(I)-Ionen und Cyanid-Ionen reagieren in wässriger Lösung zum einem weißen Niederschlag von Silber(I)-cyanid, der sich mit Cyanid-Ionen im Überschuss zum komplexen Anion Dicyanidoargentat(I) löst.
![{\displaystyle \mathrm {Ag^{+}+2\ CN^{-}\longrightarrow AgCN\downarrow +CN^{-}\longrightarrow [Ag(CN)_{2}]^{-}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/7cb27ea7cb11df52e813714c174b1411fd30d111)
- Silber(I)-Ionen und Thiocyanat-Ionen reagieren in wässriger Lösung zum einem weißen Niederschlag von Silber(I)-thiocyanat, der sich mit Thiocyanat-Ionen im Überschuss zum komplexen Anion Dithiocyanatoargentat(I) löst.
![{\displaystyle \mathrm {Ag^{+}+2\ SCN^{-}\longrightarrow AgSCN\downarrow +SCN^{-}\longrightarrow [Ag(SCN)_{2}]^{-}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/d132449898e96df0b7ea09359815eba50eda4d0a)
Nachweis mit Chromat
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Fällungsreaktion |
| pH | 7 neutral |
| Indikation | rotbrauner Niederschlag |
Durchführung
[Bearbeiten]Auch ist der Nachweis als Silberchromat charakteristisch. Das Silber(I)-Ion fällt aus neutraler Probelösung mit Chromat-Ionen als rotbraunes Silberchromat. Es ist in verdünnter Salpetersäure sowie Ammoniak löslich.
Erklärung
[Bearbeiten]- Silber(I)-Ionen und Chromat-Ionen reagieren in wässriger Lösung zum einem rotbraunen Niederschlag von Silber(I)-chromat.

Silicium
[Bearbeiten]Silicium kommt im anorganischen Praktikum als Silicat SiO32– vor.
Silicat
[Bearbeiten]Silicatanionen SiO44− bilden säureschwerlösliche Salze. In Mineralien kommt sie sehr häufig vor.
Bleitiegelprobe / Wassertropfenprobe
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Verdrängungsreaktion |
| pH | <7 schwefelsauer |
| Indikation | weißes Silicumoxid |
Durchführung
[Bearbeiten]Zum Nachweis der Silikatanionen wird eine kleine Portion der Probe in einen Bleitiegel gegeben, mit gepulverter Calciumfluorid versetzt (Mengenverhältnis Probe : Calciumfluorid etwa 3:1) und vermischt. Anschließend überschichtet man vorsichtig mit Schwefelsäure. Es bildet sich das gasförmige Siliciumtetrafluorid.
Man verschließt den Tiegel mit einer PVC-Platte, an deren Unterseite sich ein kleiner Wassertropfen befindet (dieser darf natürlich nicht in die Probe hängen) und lässt ihn ungefähr eine Minute stehen. Das SiF4 reagiert mit dem Wasser wieder zu weißem Siliciumoxid, das sich kraterförmig im Tropfen absetzt.
Eine Alternative zur PVC-Platte mit Wassertropfen ist ein schwarzes Filterpapier, das angefeuchtet wird. Das entweichende SiF4-Gas zersetzt sich dort zu SiO2, was an einem weißen Fleck erkennbar ist.
Hinweis: Keinen Überschuss von CaF2 benutzen, da sonst H2SiF6 gebildet wird.
Erklärung
[Bearbeiten]- Fluoridanionen reagieren mit Schwefelsäure zu Sulfatanionen und Fluorwasserstoff.
- Siliciumdioxid reagiert mit Fluorwasserstoff zu Silicumtetrafluorid und Wasser.

Die Hinreaktion läuft unten im Tiegel ab, die Rückreaktion oben am Deckel.
Schwefel
[Bearbeiten]Schwefel kommt im anorganischen Praktikum als Sulfat SO42–, Thiosulfat S2O32−, Sulfit SO32– und Sulfid S2– vor.
Sulfid
[Bearbeiten]Sulfid-Anionen mit Bleiacetatpapier
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Fällungsreaktion |
| pH | ? |
| Indikation | schwarze Färbung |
Sulfid-Ionen (S2−) lassen sich mit Bleiacetatpapier nachweisen, wobei eine schwarze Färbung des Papiers eintritt, hervorgerufen von Bleisulfid.
Erklärung
[Bearbeiten]- Sulfid-Ionen reagieren mit Blei(II)acetat zu Blei(II)sulfid und Acetat-Ionen.

Stinkprobe: Nachweis als Schwefelwasserstoff
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Verdrängungsreaktion |
| pH | <<7 |
| Indikation | Gestank |
Eine weitere Möglichkeit ist das Ansäuern einer festen Probe mit einer starken Säure. Es entsteht ein abscheulicher, charakteristischer Geruch nach faulen Eiern, hervorgerufen durch das Gas Schwefelwasserstoff, welches mit der Säure aus dem Sulfid verdrängt werden konnte. Das H2S-Gas hat die gleiche Toxizität wie Blausäure und sollte entsprechend unter dem Abzug gehandhabt werden.
Erklärung
[Bearbeiten]- Sulfid-Ionen reagieren mit Oxonium-Ionen zu dem Gas Schwefelwasserstoff.

Iod-Azid-Reaktion
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Redoxreaktion |
| pH | ? |
| Indikation | Entfärbung, Gasentwicklung |
Reine Lösungen von Natriumazid (NaN3) und Iod (I2) sind längere Zeit nebeneinander beständig. Sie werden aber durch Einwirkung von S2− (auch schwerlösliche Schwermetallsulfide) katalytisch zersetzt. SCN− und Thiosulfat (und auch alle entsprechenden Verbindungen mit Schwefel der Oxidationsstufe -2) reagieren analog.
Durchführung
[Bearbeiten]Reagenz: 1 g NaN3 in 75 ml Wasser bzw. 1 g I2 in 75 ml Ethanol
Auf der Tüpfelplatte wird etwas Ursubstanz oder eine kleine Menge Niederschlag mit 1 Tropfen Reagenzlösung versetzt. Die Entwicklung von freien Gasbläschen (durch Zersetzung von Azid-ionen) und gleichzeitige Entfärbung der Reaktionslösung (durch Reduktion von Iod) deuten auf Anwesenheit von S2−.
Da die eingesetzten Substanzmengen meist relativ gering sind, ist die Gasentwicklung nicht immer gut zu erkennen.
Erklärung
[Bearbeiten]- Sulfidanionen und Iod reagieren zu Schwefel und Iodid. (Entfärbung)

- Schwefel und Azidionen reagieren zu Sulfid und molekularem Stickstoff. (Gasentwicklung)

Störungen
[Bearbeiten]Größere Mengen an I− stören die Reaktion. In diesem Fall bewirkt die Zugabe von einigen Tropfen Hg(NO3)2-Lösung die Bildung von [HgI4]2−. Letzteres hat keinen Einfluss auf die beschriebene katalytische Zersetzung von Iod/Azid.
Sulfit
[Bearbeiten]Sulfit mit Permanganat
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Redoxreaktion |
| pH | <7 essigsauer |
| Indikation | Entfärbung |
Schon die Redoxreaktion mit Kaliumpermanganat als Oxidationsmittel kann einen Hinweis auf Sulfit (SO32–) geben (wie auch auf Eisen(II)-Ionen und alle anderen Reduktionsmittel). Diese Reaktion kann man sich jedoch für einen indirekten Nachweis zu Nutze machen

Durchführung
[Bearbeiten]Man gibt zu 10 ml einer essigsauren (keinesfalls schwefelsauer!) Kaliumpermanganat-Lösung (schwach rosaviolett) 10 Tropfen verdünnte Bariumchloridlösung. (siehe Bild: linkes Reagenzglas)
Die zu untersuchende Substanz wird nun dieser Reagenzlösung zügig zugegeben. Beim Verschwinden der rosavioletten Farbe und Ausfällen eines weißen Niederschlages, enthielt die Ursubstanz Sulfit-Ionen (siehe Bild: mittiges Reagenzglas).
Erklärung der Erscheinung: Kaliumpermanganat oxidiert wie oben beschrieben Sulfit-Ionen zu Sulfat-Ionen; das Permanganat selbst wird zu farblosem Mangan(II) reduziert; Folge: Entfärbung. Das entstandene Sulfat kann nun mit der Nachweisreaktion #Sulfat als Bariumsulfat bestätigt werden.
Achtung! Es kann passieren, dass die violette Farbe anfangs immer schwächer wird, ein weißer Niederschlag auch ausfällt, jedoch eine gewisse Farblichkeit bestehen bleibt und diese trotz Zugabe weiterer Ursubstanz nicht verschwindet (siehe Bild: rechtes Reagenzglas). Dann haben sich Permanganat-Ionen im regelmäßigen Kristallgitter des Bariumsulfats eingelagert. Diese sind dort fest angeordnet und können folglich nicht reduziert werden. Dieser Sachverhalt tritt hauptsächlich ein, wenn die Ursubstanz zu langsam zugegeben wurde oder die Konzentration der Sulfit-Ionen zu schwach ist. Der Vorgang sollte dann wiederholt werden.
Erklärung
[Bearbeiten]Erklärung der Erscheinung: Kaliumpermanganat oxidiert wie oben beschrieben Sulfit-Ionen zu Sulfat-Ionen; das Permanganat selbst wird zu farblosem Mangan(II) reduziert; Folge: Entfärbung.
- Sulfit-Ionen reagieren mit Permanganat-Ionen in saurer Umgebung zu Mangan(II)-Ionen, Sulfat-Ionen und Wasser.

Störung
[Bearbeiten]Um das Entstehen von Bariumcarbonat zu verhindern, muss die Reagenzlösung essigsauer sein. Bevor man den Nachweis durchführt, ist die Probelösung mit salzsaurer Bariumchloridlösung unbedingt auf Sulfat-Ionen zu prüfen. Bei Anwesenheit dieser müssen sie zuvor vollständig entfernt werden. Dies kann man wie folgt beschrieben ohne Sulfit-Verlust erreichen:
Zum Gelingen der Trennung ist zügiges Arbeiten zu verlangen. 20 mL der Probesubstanz werden mit 10 ml einer HCl/BaCl2-Lösung (c(H3O+)=1 mol/L) filtriert. (Zusammenführen der Lösungen erst kurz vor Filtrierung!) Im Erlenmeyerkolben werden 10 mL einer 0,5-molaren Natronlauge vorgelegt. Die Sulfat-Ionen werden gefällt und bleiben als Bariumsulfat im Rückstand. Entstehendes Bariumsulfit ist säurelöslich, sodass die Sulfit-Ionen ins Filtrat gelangen. Die vorhandene Natronlauge verhindert den Verlust der Sulfit-Ionen durch eventuelle Reaktion mit den Wasserstoff-Ionen der Säure. Ein Niederschlag im Filtrat durch entstehendes Bariumhydroxid kann in einem sauberen Filter abfiltriert werden. Jetzt kann die Lösung auf Sulfit-Ionen geprüft werden.
Sulfit mit Schwefelsäure
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Verdrängungsreaktion |
| pH | <7 essigsauer |
| Indikation | Schwefeldioxidgas |
Durchführung
[Bearbeiten]Sulfit-Ionen (SO32−) lassen sich auch per Verdrängungsreaktion mit (konzentrierter) Schwefelsäure nachweisen. Es entsteht ein stechender Geruch von Schwefeldioxid, der mittels feuchtem Unitest-Papier nachgewiesen werden kann:
Erklärung
[Bearbeiten]- Sulfit-Ionen reagieren mit Schwefelsäure zu Schwefeldioxid, Wasser und Sulfat-Ionen.

Sulfat
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Fällungsreaktion |
| pH | <7 leicht salzsauer |
| Indikation | farbloser Niederschlag |
Sulfat (SO42–) lässt sich durch Fällung als Bariumsulfat nachweisen.
Durchführung
[Bearbeiten]Dazu wird die leicht mit Salzsäure (HCl) angesäuerte Probenlösung mit einigen Tropfen Bariumchloridlösung (BaCl2) versetzt. Ist Sulfat vorhanden, so fällt unmittelbar ein farbloser, feinkristalliner Niederschlag aus.
Erklärung
[Bearbeiten]
Störung
[Bearbeiten]Dieser Nachweis kann durch Vorliegen von F–-Ionen gestört werden. In diesem Fall kann sich BaF2 bilden, welches ebenfalls ausfällt. Dieses geht allerdings beim Erhitzen mit Salzsäure wieder in Lösung. Falls nicht angesäuert wird, können andere schwerlösliche Bariumsalze ausfallen.
Modifikation: Nachweis als Bariumsulfat-Kaliumpermanganat-Mischkristall
[Bearbeiten]Wird die Probelösung wie oben beschrieben mit Bariumchlorid und zusätzlich mit verdünnter Kaliumpermanganatlösung (KMnO4) versetzt, so bildet sich ein blassrosa Mischkristall, BaSO4 · KMnO4. Dieser lässt sich durch Zugabe von Wasserstoffperoxidlösung (H2O2) nicht entfärben.
Thiosulfat
[Bearbeiten]Sonnenuntergangsreaktion
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Fällungsreaktion, Disproportionierung (Redox) |
| pH | <7 leicht salzsauer |
| Indikation | farbloser Niederschlag, dann über gelb, orange, braun zu schwarz |
Thiosulfat-Anionen (S2O32−) werden durch Zugabe von Silbernitratlösung im Überschuss bei pH um 7 nachgewiesen („Sonnenuntergang“): Es entsteht ein farbloser Niederschlag, der sich langsam von gelb, orange über braun bis hin zum schwarzen Silbersulfid verfärbt (Reaktion in 2 Schritten – Ausfällung mit anschließender Redoxreaktion in Form einer Disproportionierung).
Wird die Lösung eisgekühlt, lässt sich der Farbwechsel besser verfolgen.
Erklärung
[Bearbeiten]- 1.Schritt: Thiosulfat wird durch Silberionen schnell ausgefällt, es entsteht sofort weißes Silberthiosulfat.

- 2. Schritt: Das Thiosulfat (Oxidationszahl Schwefel: +II) zerfällt langsam in Verbindungen mit günstigerer Oxidationszahl: Sulfid (−II) und Sulfat (+VI).

Thiocyanat
[Bearbeiten]Stierblutprobe
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Komplexbildung |
| pH | |
| Indikation | tiefrote Färbung |
Thiocyanat-, oder auch Rhodanid-Ionen werden qualitativ mit der „Stierblutprobe“ nachgewiesen. Diese Reaktion wird auch zum Nachweis von Eisen mit Thiocyanatlösung eingesetzt.
Durchführung
[Bearbeiten]Dabei wird der zu untersuchenden Lösung eine gesättigte Eisen(III)-chlorid-Lösung zugegeben. Erscheint eine intensiv „stierblutrote“ Färbung, so waren Thiocyanat-Ionen vorhanden.
Erklärung
[Bearbeiten]- Reaktion: Thiocyanat-Ionen und Eisen(III)-Ionen reagieren im wässrigen Milieu zum Komplex Pentaaquathiocyanatoferrat(III), welcher blutrot erscheint.
Nachweis mit Kupfersulfat
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Redoxreaktion, Komplexbildung |
| pH | |
| Indikation | zunächst grün, im Überschuss schwarz, mit Sulfit weiß |
Ein weiterer spezifischer Nachweis kann mit Kupfersulfatlösung erfolgen.
Durchführung
[Bearbeiten]Zur in Wasser gelösten Ursubstanz wird frisch bereitete Kupfer(II)-sulfat zugegeben. Bei Reaktion von Thiocyanat-Ionen mit Kupfer(II)-Ionen beobachtet man zunächst eine grüne Färbung der Lösung (siehe Bild linkes Reagenzglas). Beim Vorhandensein von Thiocyanat-Ionen oder Kupfer(II)-Ionen im Überschuss bildet sich ein schwarzer Niederschlag. (siehe Bild mittiges Reagenzglas) Wird der Niederschlag mit Sulfit-Ionen behandelt, so löst sich der schwarze Niederschlag und es bildet sich ein weißer NS. (Redoxreaktion, siehe Bild rechtes Reagenzglas)
Erklärung
[Bearbeiten]Reaktion: Thiocyanat-Ionen reagieren mit Kupfer(II)-Ionen zu schwarzem, wasserunlöslichem Kupfer(II)-thiocyanat.
Reaktion: Schwarzes Kupfer(II)-thiocyanat reagiert mit Sulfit-Ionen im wässrigen Milieu zu weißem Kupfer(I)-thiocyanat, Thiocyanat-Ionen, Sulfat-Ionen und Oxonium-Ionen.
Vanadium
[Bearbeiten]Eine Vorprobe liefert die Phosphorsalzperle, bei der Vanadium in der Reduktionsflamme charakteristisch heiß bräunlich und kalt grün erscheint. Die Oxidationsflamme ist heiß rotbraun und kalt: orange.
Nachweis als Sulfid
[Bearbeiten]Mit Ammoniumsulfid erfolgt aus neutraler oder basischer Lösung keine Fällung. Es bilden sich stattdessen lösliche braun bis rotviolette Thiovanadate. Beim Sättigen der Lösung mit Schwefelwasserstoff beobachtet man eine Rotviolettfärbung durch [VS4]3-. Durch Versetzen mit Säure fällt braunes V2S5 aus.

Nachweis mit Wasserstoffperoxid
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Oxidationsreaktion |
| pH | <7 sauer |
| Indikation | rötlich-braun bis gelb |
In saurer Lösung entsteht mit Wasserstoffperoxid zunächst das rötlich-braune [V(O2)]3+, aus dem sich bei weiterem Peroxidzusatz gelb gefärbte Peroxovanadinsäure [VO2(O2)2]3- bzw. H3[VO2(O2)2] bildet.
![{\displaystyle \mathrm {VO_{4}^{3-}+H_{2}O_{2}+6\ H_{3}O^{+}\longrightarrow [V(O_{2})]^{3+}+10\ H_{2}O} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/589e99ddb4ccb352b552d8c24eb68e1e185adee6)
![{\displaystyle \mathrm {VO_{4}^{3-}+2\ H_{2}O_{2}\longrightarrow [HVO_{2}(O_{2})_{2}]^{2-}+OH^{-}+H_{2}O} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/387979172970bb46b054f107f35d6b96273b1ddb)
Störung
[Bearbeiten]Titan(IV) gibt eine analoge Reaktion und muss daher vorher abgetrennt werden.
Dichromat und Vanadat lassen sich nebeneinander nachweisen, da sich Dichromat mit organischen Lösungsmitteln ausschütteln lässt, während Vanadat in der wässrigen Phase verbleibt. Aufpassen: bei zu niedrigem pH zerfällt CrO5.
Zink
[Bearbeiten]Nachweis als Zinksulfid
[Bearbeiten]_reactions.JPG)
| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Fällungsreaktion |
| pH | 7 neutral |
| Indikation | weißer voluminöser Niederschlag |
Zink-Kationen können mit einer Alkalisulfidlösung oder einer konz. Schwefelwasserstofflösung nachgewiesen werden. Eigentlich handelt es sich um eine Sulfidfällung im Kationentrennungsgang, die Reaktion ist aber spezifisch für Zink-Ionen, da Zinksulfid das einzige schwerlösliche Sulfid ist, das eine weiße Farbe hat. Er wird jedoch durch alle anderen dunkleren Sulfidniederschläge verdeckt, so dass diese vorher abgetrennt werden müssen.
Durchführung
[Bearbeiten]Die Fällung von Zinksulfid kann etwas kniffliger werden. Es sollte im neutralen pH-Bereich gefällt werden, da Zinksulfid schon in verdünnten Mineralsäuren löslich ist. Eine Fällung mit Ammoniumacetat gepufferter Essigsäure und Ammoniumsulfidlösung sei hier empfohlen. Falls man in der Ammoniumsulfidgruppe auf Schwierigkeiten stößt, kann man auch versuchen aus dem Kaliumhydroxidauszug zu arbeiten.
Erklärung
[Bearbeiten]- Zink-Kationen reagieren mit Sulfid-Ionen zum weißen, schwerlöslichen Zinksulfid

Nachweis mit gelben Blutlaugensalz
[Bearbeiten]-Ionen_mit_Blutlaugensalze.jpg)
| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Fällungsreaktion |
| pH | 7 neutral |
| Indikation | schmutzig weißer Niederschlag |
Durchführung
[Bearbeiten]Einige Tropfen einer salzsauren, mit Acetat gepufferten Lösung werden mit wenigen Tropfen verdünnter Kaliumhexacyanidoferrat(II)-Lösung versetzt. Es entsteht ein schmutzig weißer Niederschlag, der sich in der Wärme bildet und sich in konzentrierter Salzsäure sowie verdünnter Natronlauge wieder löst. Der Niederschlag ist auf einer dunklen Tüpfelplatte am besten sichtbar.
Erklärung
[Bearbeiten]- Zink(II) reagiert mit Kaliumhexacyanoferrat(II) zu Kaliumzinkhexacyanidoferrat(II)
![{\displaystyle \mathrm {3\ Zn^{2+}+2\ K^{+}+2\ [Fe(CN)_{6}]^{4-}\longrightarrow K_{2}Zn_{3}[Fe(CN)_{6}]_{2}\downarrow } }](https://wikimedia.org/api/rest_v1/media/math/render/svg/8e4a6f23a30c6bfa4e61d518a3f68aa78672e9ed)
Nachweis als Rinmans Grün
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Bildung eines Gemisches von ZnO und CoO |
| pH | egal |
| Indikation | grüne Rinne |
Dieser Nachweis kann als Vorprobe aus der Ursubstanz oder aus dem Trennungsgang durchgeführt werden.
Durchführung
[Bearbeiten]Man gibt Analysensubstanz und wenige Tropfen einer stark verdünnte Cobaltnitrat-Lösung auf eine Magnesiarinne und glüht diese kurz in der oxidierenden Flamme des Bunsenbrenners. Arbeitet man mit einer Lösung aus dem Kationentrennungsgang, so stippt man die Rinne zuerst in die Cobaltnitratlösung und pipettiert dann etwas Analysenlösung darauf. Ist Zink in der Lösung so bildet sich Rinmans Grün. Ist die Rinne schwarz gefärbt so war zu viel Cobaltnitrat im Gemisch.
Erklärung
[Bearbeiten]- Zink(II) reagiert mit Cobaltnitrat zu einer grünen Zink-Cobalt-Spinellverbindung

Zinn
[Bearbeiten]Zinn fällt im Kationentrennungsgang in der Arsengruppe aus.
Nachweis mittels Leuchtprobe
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Redoxreaktion |
| pH | <7 salzsauer |
| Indikation | blaue Lumineszenz |


Die Leuchtprobe ist ein empfindlicher Nachweis für Zinn(II)-Ionen. Sie kann als Vorprobe oder als Nachweis im Kationentrennungsgang erfolgen.
Durchführung
[Bearbeiten]Die zu prüfende feste Substanz wird mit etwas festem Zink und 20-prozentiger Salzsäure vermischt. Nachdem man 15 Minuten gewartet hat, füllt man ein Reagenzglas mit kaltem Wasser oder Eis. Jetzt stippt man das Reagenzglas mit der Außenseite in die Mischung aus Zink, Salzsäure und Analysensubstanz und hält es in eine entleuchtete Bunsenbrennerflamme. Sieht man am Rand des Reagenzglases eine blaue Lumineszenz, so war Zinn in der Probe.
Die blaue Lumineszenz kann man leicht mit der blauen Bunsenbrennerflamme verwechseln, deshalb sollte man vorher eine Vergleichsprobe mit einer zinnhaltigen Substanz und eine Blindprobe ohne Zinn gemacht haben. Auf der anderen Seite kann das Leuchten auch schwer zu erkennen sein. Tipp: Statt Wasser, eine dunkel gefärbte Kaliumpermanganatlösung in das Reagenzglas füllen, damit man einen besseren Kontrast hat. Ein abgedunkelter Abzug hilft auch die Lumineszenz besser zu erkennen.
Erklärung
[Bearbeiten]Hält man das Reagenzglas in die Bunsenbrennerflamme, so entsteht die blaue Lumineszenz, zu deren Ursprung es verschiedene Meinungen gibt. Es entsteht entweder durch das Gas Stannan oder Zinnchloride in verschiedenen Oxidationsstufen.[1]
Bei der Zugabe von Salzsäure zu Zink entsteht naszierender Wasserstoff, ein sehr gutes Reduktionsmittel.
- Zink und Salzsäure reagiert zu naszierendem Wasserstoff und Zinkchlorid.

Das Gas Stannan sorgt für das Leuchten
- Zinn(II)-Ionen reagieren mit naszierendem Wasserstoff zu Stannan.

Das Zink reduziert eventuell vorhandene schwerlösliche Sn(IV)-Verbindungen in Sn(II)-Verbindungen:

Es entsteht durch Zinn(II)-chlorid.

Es entsteht direkt Zinn(IV)-chlorid.

Störung
[Bearbeiten]Niob bildet ähnlich fluoreszierende Verbindungen und führt zu falsch positiven Nachweisen. Größere Mengen Arsen stören ebenfalls, da AsH3 auch mit blauer Flamme brennt.
Nachweis als Molybdänblau
[Bearbeiten]| Nachweisreaktion | |
|---|---|
| Reaktionstyp: | Redoxreaktion |
| pH | 7 |
| Indikation | blaue Ringe |
Durchführung
[Bearbeiten]Zur Probe auf Zinn wird die Probelösung mit Zinkperlen versetzt um Zinn(IV) zu Zinn(II) zu reduzieren. Auf ein mit 5-prozentiger Ammoniummolybdat-Lösung getränktes Filterpapier wird die Lösung pipettiert. Es entstehen blaue Ringe (Molybdänblau, CAS 66771-43-5), die beim Trocknen des Filterpapiers deutlicher zu sehen sind und Zinn anzeigen.
Erklärung
[Bearbeiten]- Molybdatlösung reagiert mit Zinn(II)-Kationen zu Molybdänblau und Zinnoxid

- ↑ Zum Nachweis von Zinn (II) DOI:10.1007/BF00533516
Kationentrennungsgang
[Bearbeiten]
Salzsäuregruppe
[Bearbeiten]
Schema der Salzsäuregruppe

Fällung und Filtration der Salzsäuregruppe
[Bearbeiten]Bei einem pH-Wert von 0 bis 3,5 fallen in HCl-haltiger Lösung die weißen/farblosen Chloride von Ag(I), Pb(II) und Hg(I/II) aus:
- Quecksilber(I)-chlorid – Hg2Cl2
- Quecksilber(II)-chlorid – HgCl2
- Blei(II)-chlorid – PbCl2 - löslich in heißem Wasser
- Silberchlorid – AgCl – als Silberdiamminkomplex löslich in konz. Ammoniakwasser. Ist die Konzentration der Salzsäure zu hoch, kann das AgCl komplexiert werden und geht dann als [AgCl2]– in Lösung. Bei Verdacht auf Silber sollte die Lösung u.U. etwas mit Wasser verdünnt werden. AgCl fällt dann aus.
Abtrennung des Blei(II)-chlorides
[Bearbeiten]Der Niederschlag wird unter dem Abzug mehrmals mit 1 ml Wasser und 1 Tropfen ca. 2-molarer Salzsäure aufgekocht und heiß durch einen durch kochendes Wasser oder im Trockenschrank und mit Filterpapier auf rund 100 Grad Celsius vorgewärmten Glastrichter filtriert. Das Filtrat lässt man abkühlen, um hieraus Blei durch Zugabe von einem Tropfen ca. 0,5-molarer Kaliumchromat- oder Kaliumdichromatlösung als gelbes Blei(II)-chromat PbCrO4 nachzuweisen.
Alternativ kann Natriumiodidlösung zugegeben, mit der Blei(II)-Ionen als gelbes Blei(II)-iodid PbI2 ausfallen. Alternativ kann man auch eine Nadel auskristallisierendes Blei(II)-chlorid nehmen und diese auf ein Kaliumiodid-Papier legen. Da sich das schwerlöslichere gelbe Bleiiodid bildet, ist nach einigen Minuten um die Nadel herum ein gelber Hof von Blei(II)-Iodid zu sehen.
Trennung des Quecksilber(I)-chlorides (Hg2Cl2) vom Silberchlorid
[Bearbeiten]Ein Teil des Filterrückstandes wird im Reagenzglas mit 1 ml konz. Ammoniaklösung behandelt. Das Auftreten einer Schwarzfärbung (durch Bildung u.a. von Quecksilber) beweist die Gegenwart von Quecksilbersalzen. Der schwarze Niederschlag wird abfiltriert und das Filtrat mit einem Tropfen Salzsäure angesäuert. Es entsteht unter Umständen ein weißer Niederschlag von Silberchlorid, der sich bei Zugabe von verdünntem Ammoniakwasser unter Bildung des Diammin-Silber(I)-komplexes [Ag(NH3)2]+ löst.
![{\displaystyle \mathrm {Hg_{2}Cl_{2}+NH_{3}\longrightarrow Hg^{0}\downarrow +[Hg(NH_{2})]Cl\downarrow +HCl} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/187cc32b98a50b4611cdde7fd23787dac7d72e0c)
![{\displaystyle \mathrm {AgCl+2\ NH_{3}\longrightarrow [Ag(NH_{3})_{2}]Cl} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/696a524e55fe5440e47fd368e4c8d4da4840e039)
Schwefelwasserstoffgruppe
[Bearbeiten]Die Gruppenfällung mit Schwefelwasserstoff
[Bearbeiten]Bei einem pH-Wert von 0 bis 5 fallen in H2S-haltiger Lösung die Sulfide von As(III,V), Sb(III,V), Sn(II,IV), Hg(II), Cu(II), Pb(II), Bi(III,V) und Cd aus. Die zu analysierende Lösung muss zu Beginn der Fällung schwach salzsauer bis essigsauer sein. Ist die Analyse in konzentrierten oxidierenden Säuren gelöst worden, muss dieses Oxidationsmittel komplett durch Sieden vertrieben werden, da sich sonst während der Zugabe von H2S eventuell S8 bilden könnte. Bei gleichen Mengen der Stoffe in der Analysesubstanz fallen die Sulfide dann in folgender Reihenfolge aus:
- As2S3 (gelb)
- SnS2 (hellgelb)
- Sb2S5 (orange)
- HgS (schwarz)
- PbS (schwarz)
- CuS (schwarz)
- SnS (schwarz bis braun)
- Bi2S3 (schwarz bis braun)
- zuletzt CdS (eigelb)
Da H2S ein sehr giftiges Gas ist, wird stattdessen oft mit einem Gemisch von Na2S mit NH4Cl/HCl Puffer (alles aq) gearbeitet. Eine weitere Möglichkeit ist 5 ml Ammoniumsulfid in 80 ml verdünnte Salzsäure einzurühren. Nach einer Stunde sinkt die ~0.15 molare Lösung auf 0.1 molar ab.
- Ammoniumsufild und Salzsäure reagieren zu Schwefelwasserstoff und Ammoniumchlorid

Achtung: Auch wenn hier mit wässrigen Lösungen gearbeitet wird, bilden sich ständig giftige Schwefelwasserstoffdämpfe. Das H2S-Wasser nur unter dem Abzug und am besten in einem abgedeckelten Gefäß aufbewahren.
Zunächst werden aus relativ saurer Lösung (niedriger pH) die schwerlöslichen Sulfide gefällt. Dann wird die Lösung verdünnt und bei max. pH 3,5 die weniger schwerlöslichen, aber noch im Sauren fällbaren Sulfide gefällt. Der pH Wert ist ständig zu kontrollieren, besonders wenn der pH-Wert nicht nur durch weitere Zugabe der Na2S-Lösung (hier Na2S-Lösung=H2S Wasser) erhöht wird.
(Hinweise: Logarithmische Skala! In der Praxis wird häufig zusätzlich NH3 dazu gegeben. Dies wird jedoch als unsaubereres Arbeiten betrachtet! Wird der pH-Wert nicht eingehalten fällt unter Umständen CdS nicht, bzw. die Sulfide der nachfolgenden Gruppe (NH4)2S-Gruppe fallen zu früh aus. Hier ist das Löslichkeitsprodukt zu beachten!)
Man verfährt mithin z.B. folgendermaßen: Das Filtrat der Salzsäuregruppe wird bis fast zur Trockene eingedampft (Befreiung von Salpetersäure), mit etwa 0,5 mL halbkonz. Salzsäure unter Erwärmen gelöst und in ein Normal-Reagenzglas überführt. Durch eine Kapillarpipette wird etwa 3 min Schwefelwasserstoff eingeleitet (oder es werden unter dem Abzug 1-2 mL Ammoniumsulfid-Lösung zugegeben, wobei der - ggf. gepufferte - pH-Wert unter 4-5 bleiben muss!).
Nach einer Minute wird die Lösung auf etwa das Fünffache verdünnt - also max. 10 mL. Danach wird erneut der pH-Wert kontrolliert, gegebenenfalls wieder angesäuert und filtriert. Der Filterrückstand wird - immer noch unter dem Abzug - mit Schwefelwasserstoff-Lösung (ca. 1 mol/L) gewaschen. In einen halben mL des mit dem Waschwasser vereinigten Filtrates wird erneut Schwefelwasserstoff-Wasser gegeben. Wenn dabei nichts mehr ausfällt (pH stets unter 7 halten!), war die Fällung quantitativ - ansonsten ist das Einleiten und Filtrieren zu wiederholen.
(Hinweise: Zur Kontrolle kann ein kleiner Teil des Filtrats auch mit Cd-Acetat versetzt werden. Fällt sofort das typische gelbe Kadmiumsulfid CdS aus,so ist der S2- Gehalt der Analyselösung hoch genug und man kann annehmen das alle Sulfide die in der Analyse enthalten sind bereits gefallen sind. Bei Verwendung der Na2S-Lösung entsteht mitunter auch rotes HgS. As(V), Sb(V), Bi(V) und Sn(IV) oxidieren S2- zu elementarem Schwefel. Dieser schwimmt häufig oben auf der Lösung auf oder bleibt sogar nach dem Filtrieren kolloid in Lösung).
Trennung der H2S Gruppe in 2 Untergruppen
[Bearbeiten]Zunächst muss die Arsen- von der Kupfergruppe getrennt werden:
Der Niederschlag (Nd.) der Schwefelwasserstoff-Gruppe wird im Reagenzglas mit 2 mL gelber Ammoniumpolysulfid-Lösung bei etwa 50-60°C ausgelaugt (auslaugen = Feststoffgemisch zwecks teilweiser Auflösung desselben unter Umrühren in Flüssigkeit erwärmen), danach filtriert. Der Filterrückstand ist mit 1-2 mL Wasser auszuwaschen: Im Filtrat findet sich die As-Sn-Gruppe (lösliche Thiosalze: AsS43– SbS43– SnS32-), im Filterrückstand die Cu-Gruppe.
- z.B.

Sodann wird Schwefel unter Sieden und Rühren im Ammoniumsulfidlösung gelöst. Es bildet sich gelbe Ammoniumpolysulfidlösung. Die Sulfide von As, Sb und Sn (Arsen-Gruppe) bilden also mit dieser Ammoniumpolysulfidlösung (NH4)2Sx- bzw. mit LiOH/KNO3- Lösung lösliche Thiometallat- bzw. Oxothiometallat-Komplexe. Die Sulfide von Hg, Pb, Bi, Cu und Cd (Kupfer-Gruppe) lösen sich nicht und bleiben zurück. Die Sulfidfällung ist mit ausreichend H2S Wasser zu waschen, solange bis kein Cl– mehr im Waschwasser ist. (Nachweis: AgNO3 Lösung zum Waschwasser tropfen). Danach den Niederschlag (Nd.) mit (NH4)2Sx digerieren.
- z.B.

Kupfergruppe, Abtrennung von Quecksilberionen
[Bearbeiten]Der Nd. der Cu-Gruppe wird mit 1-2 mL warmer halbkonzentrierter HNO3 (ca. 4-7 mol/L Salpetersäure) erwärmt. Es lösen sich alle Sulfide bis auf HgS.
(Hinweise: Proben auf Quecksilber durchführen, z.B. "Amalgamprobe" mit einem Stück Kupferblech, Zur Sicherheit HgS in wenig konz. HCL/ konz HNO3, lösen HNO3 abrauchen und weitere Hg Proben durchführen. Reduktion mit SnCl2 usw.)
Abtrennung von Blei(II)-Ionen
[Bearbeiten]Das salzsaure Filtrat vom HgS-Rückstand wird unter Zusatz von 0,5 mL konz. Schwefelsäure im Porzellanschälchen eingedampft, bis dass weiße Nebel entstehen (Schwefeltrioxid). Nach dem Abkühlen wird vorsichtig mit verdünnter Schwefelsäure verdünnt: In Gegenwart von Blei bildet sich ein weißer Niederschlag (PbSO4 Blei-II-Sulfat). Dieser wird abfiltriert, in Ammoniumtartratlösung gelöst (Komplexbildungsreaktion) und wie in der HCl-Gruppe nachgewiesen mit Iodid-, Natriumsulfid-, Kaliumdichromat-Lösung.
Bismut sowie Kupfer neben Cadmium nachweisen
[Bearbeiten]
Die Lösung ist nun ammoniakalisch zu machen. Kupfer ist sofort an der blauen Farbe der Lösung zu erkennen:
- Kupfer(II)-Kationen reagieren mit Ammoniak zu dem blauem Tetraaminkupfer(II)-Komplex.
![{\displaystyle \mathrm {Cu^{2+}+4\ NH_{3}\longrightarrow [Cu(NH_{3})_{4}]^{2+}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/6d37f99210a906f5833072eb5d2ba02dd91bb36b)
(Achtung die Färbung kann auch sehr schwach sein! NH3 im Überschuss zugeben, bei Anwesenheit von Bi fällt weißes Bi(OH)3. Dieses löst sich in HCl. Zum Nachweis Niederschlag neutralisieren und mit alkalischer Stannat(II) Lsg. versetzten (Elementares Bismut fällt schwarz aus) oder mit Natriumiodidlösung (zunächst fällt schwarzes Bismut-III-iodid aus, das sich dann im Iodidüberschuss als orangefarbiger Tetraiodobismutat-Komplex löst).
Nach Zugabe von KCN zur Hauptlösung der Kupfergruppe muss sich die Lösung entfärben (ein zusätzlicher Nachweis für Cu).
- Der blaue Kupfertetraminkomplex reagiert mit Cyanid zu einem Kupfer(I)tetracyanido-Komplex und dem giftigen Gas Dicyan
![{\displaystyle \mathrm {2\ [Cu(NH_{3})_{4}]^{2+}+10\ CN^{-}\longrightarrow 2\ [Cu(CN)_{4}]^{3-}+(CN)_{2}\uparrow } }](https://wikimedia.org/api/rest_v1/media/math/render/svg/329ef4b6fc638f3e2d455988f54211045f67918d)
Achtung: Ab hier die Lösung nicht mehr ansäuern, sonst entsteht hochgiftige Blausäure (HCN-Gas)! Bei der Entsorgung beachten – mit konz. Wasserstoffperoxid entgiften!).
Wenn man bis zur vollständigen Entfärbung KCN zugegeben hat, kann man dann mit H2S-Wasser Cadmium als gelbes Cadmiumsulfid CdS ausfällen, ohne dass schwarzes Kupfer-II-sulfid stört (Entsteht dennoch ein dunkler Nd., so hat die Trennung innerhalb der Gruppe versagt. Falls Unsicherheiten aufgetreten sind, ist die gesamte Trennung der Kupfer-Gruppe zu wiederholen).
Arsen-Zinn-Gruppe
[Bearbeiten]Im vereinfachten Kationentrenngang in Abwesenheit giftigen Arsens verfährt man folgendermaßen:
Trennung Sb von Sn
[Bearbeiten]Die As-Sn-Gruppe (Filtrat der Cu-Gruppe, s.o.) wird unter dem Abzug mit 7M HCl kurz aufgekocht und durch mehrmaliges Filtrieren durch den gleichen Filter weitgehend von kolloidalem Schwefel befreit.
Durchführung der Nachweisreaktionen für Zinn und Antimon
[Bearbeiten]Das Filtrat wird durch unedle Metalle (Mg-, Al-, Zn-, Fe-Pulver) reduziert, so dass schwarzes, elementares Sb ausfällt (zum Einzelnachweis im Filter waschen, in Königswasser lösen, abrauchen und mit Ammoniumsulfid-Lösung als orangefarbenen Antimon-V-sulfid-Nd. nachweisen) und das Sn(IV) zu Sn(II) reduziert wird (zum Einzelnachweis Sn per Leuchtprobe s.u.)
Abtrennung von Arsen und Nachweisreaktionen für Zinn und Antimon
[Bearbeiten]Im um Arsen erweiterten Trennungsgang verfährt man hier anders:
Die Lösung der Thiometallat- bzw. Oxothiometallat-Komplexe von As, Sb, Sn ist zunächst mit HCl anzusäuern (Achtung: Schwefelwasserstoff entweicht!): Die Sulfide von As, Sb und Sn fallen erneut aus (die Farben erneut beobachten!). Die Sulfide sind nun abzutrennen und mit wenig konz. HCl zu erwärmen: As2S5 bleibt als gelbes Sulfid zurück, Sb und Sn gehen in Lösung. Mit NH3/H2O2 lässt sich das Arsensulfid in Lösung bringen und danach getrennt nachweisen, z.B. durch die Marshsche Probe.
Die Lösung ist danach einzuengen um Sb und Sn anschließend nebeneinander nachzuweisen (Nagelprobe: Einen Eisennagel in die Lösung legen, an ihm bildet sich elementares Antimon (Sb). Diese kann erneut im Sauren gelöst und mit H2S Wasser als orangefarbenes Sulfid gefällt werden. Sn verbleibt in der Lösung. Es kann sehr gut mit der Leuchtprobe nachgewiesen werden.
Ammoniumsulfidgruppe
[Bearbeiten]-
 CoNO3 (schwach pink), CoS (schwarz), Co(OH)2 (rotbraun), Cobaltcarbonate (blau)
CoNO3 (schwach pink), CoS (schwarz), Co(OH)2 (rotbraun), Cobaltcarbonate (blau) -
![NiNO3 (grün), NiS (schwarz), Ni(OH)2 (grün), [Ni(NH3)6]2+ (blau), Nickelcarbonate (grün)](//upload.wikimedia.org/wikipedia/commons/thumb/8/80/DSC01973_-_Nickel_%28II%29_reactions.JPG/85px-DSC01973_-_Nickel_%28II%29_reactions.JPG) NiNO3 (grün), NiS (schwarz), Ni(OH)2 (grün), [Ni(NH3)6]2+ (blau), Nickelcarbonate (grün)
NiNO3 (grün), NiS (schwarz), Ni(OH)2 (grün), [Ni(NH3)6]2+ (blau), Nickelcarbonate (grün) -
 FeS (schwarz), Fe(OH)2 (weiß), Fe(OH)3 (braun), FeCO3 (eigentlich weiß)
FeS (schwarz), Fe(OH)2 (weiß), Fe(OH)3 (braun), FeCO3 (eigentlich weiß) -
 MnS (orangegelb), MnO(OH) (bräunlich), MnCO3 (bräunlich, natürliches Mineral rosa)
MnS (orangegelb), MnO(OH) (bräunlich), MnCO3 (bräunlich, natürliches Mineral rosa) -
 links: Al(OH)3 (weiß), rechts: Al2(CO3)3 (weiß)
links: Al(OH)3 (weiß), rechts: Al2(CO3)3 (weiß) -
![Cr(NO3)3 (bläulich), Cr(OH)3 in gelbem (NH4)2S, Cr(OH)3 (graugrün), [Cr(OH)6]3- (grünlich), [Cr(NH3)6]3+ (gelblich), Cr(NO3)3/Cr(OH)3 (graugrün)](//upload.wikimedia.org/wikipedia/commons/thumb/5/5a/DSC01967_-_Chromium_reactions.JPG/119px-DSC01967_-_Chromium_reactions.JPG) Cr(NO3)3 (bläulich), Cr(OH)3 in gelbem (NH4)2S, Cr(OH)3 (graugrün), [Cr(OH)6]3- (grünlich), [Cr(NH3)6]3+ (gelblich), Cr(NO3)3/Cr(OH)3 (graugrün)
Cr(NO3)3 (bläulich), Cr(OH)3 in gelbem (NH4)2S, Cr(OH)3 (graugrün), [Cr(OH)6]3- (grünlich), [Cr(NH3)6]3+ (gelblich), Cr(NO3)3/Cr(OH)3 (graugrün) -
 weißes ZnS in gelbem (NH4)2S, Zn(OH)2 (weißlich), Zinkcarbonate (weißlich)
weißes ZnS in gelbem (NH4)2S, Zn(OH)2 (weißlich), Zinkcarbonate (weißlich)
_Reactions.JPG)
_reactions.JPG)

![Cr(NO3)3 (bläulich), Cr(OH)3 in gelbem (NH4)2S, Cr(OH)3 (graugrün), [Cr(OH)6]3- (grünlich), [Cr(NH3)6]3+ (gelblich), Cr(NO3)3/Cr(OH)3 (graugrün)](/wiki/Datei:DSC01967_-_Chromium_reactions.JPG)
Die Gruppenfällung mit Ammoniumsulfidlösung
[Bearbeiten]Bei einem pH-Wert von 8 bis 9 fallen in (NH4)2S -haltiger Lösung die Sulfide von Co(II), Ni(II), Mn(II), Zn(II), Eisen(II,III) – letzteres als FeS (Redoxreaktion mit Sulfidanionen als Reduktionsmittel für Eisen-III-Kationen). Als unlösliche Hydroxide fallen Al(OH)3 und Cr(OH)3 aus.
- Nickel fällt als schwarzes Nickel(II)-sulfid.

- Cobalt fällt als schwarzes Cobalt(II)-sulfid.

- Mangan fällt als rosafarbenes Mangan(II)-sulfid.

- Zink fällt als weißes Zink(II)-sulfid.
- Eisen(III) wird zu hellbraunem Eisen(II)-sulfid reduziert. Dabei entsteht elementarer Schwefel.

Bei der Ausfällung der (NH4)2S -Gruppe bilden sich neben diesen Sulfidniederschlägen auch Ausfällungen von Aluminium- und Chromhydroxid. Die Ursache zeigt sich beim Vergleich der Löslichkeitsprodukte zu Ammoniumsulfidgruppe.
Das Filtrat der Schwefelwasserstoffgruppe wird dazu in einer Porzellanschale oder einem kleinen Becherglas unter Zusatz von 1 Spatelspitze festem Ammoniumchlorid auf ca. 1 mL eingeengt. Bis zur deutlich alkalischen Reaktion wird konz. Ammoniak zugeträufelt, mit 1-2 mL Ammoniumsulfidlösung versetzt und der Nd. einige min. erwärmt, dann abfiltriert. 1 Tropfen Filtrat wird mit 1 Tropfen Blei-II-acetat versetzt (oder auf Bleiacetatpapier gegeben). Schwarzes PbS zeigt Vollständigkeit der Fällung an. Der Nd. wird sofort mit stark verdünnter Ammoniumsulfidlösung gewaschen.
(Hinweis: Bei orangeroter oder violetter Farbe zuvor mit Ethanol kochen! - Filtrat prüfen: Bei gelbbrauner Trübung durch NiS-Kolloid mit Ammoniumacetat und Filterpapierschnipseln kochen und neu filtrieren. Das wird u.U. erforderlich, denn NiS und CoS bilden Kolloide; Kolloide sind große Molekülverbände, wobei die Moleküle nur durch intermolekulare Kräfte zusammengehalten werden (Dipol, Wasserstoffbrücken). Durch ihre molekulare Struktur (hydrophile und lipophile Bereiche der Seifenmoleküle) oder durch elektrische Ladungen werden diese Molekülaggregate in Lösung gehalten (kolloidale Lösung; Thyndall-Effekt). Das Erwärmen der Lösung mit Filterpapierschnitzeln führt zu einer Vergrößerung der Teilchen, die man filtrieren kann. Durch Zugabe von Ammoniumacetat kann die elektrische Ladung aufgehoben werden und die Aggregate fallen aus).
Alkalischer Sturz
[Bearbeiten]Abtrennung und Nachweis von Cobalt und Nickel
[Bearbeiten]Der Sulfidniederschlag wird sofort mit 1-2 mL verdünnter Salzsäure behandelt, um anschließend den ungelösten Rest (CoS, NiS) abzufiltrieren und in je einigen mL verdünnter Essigsäure und konz. Wasserstoffperoxidlösung zu lösen. Danach ist aus der essigsaueren Co-Ni-Lösung Fe-Reste mit Ammoniak auszufällen, abzufiltrieren und vom Fe-Rest befreites Filtrat für die einzelnen Nachweisreaktionen von Co und Ni zu nutzen.
Abtrennung von Eisen(III)-hydroxid und Braunstein (Mangandioxid)
[Bearbeiten]Von Ni & Co befreites Filtrat der Sulfide aufkochen, konz. Salpetersäure zugeben und eindampfen, neutralisierte Rest-Tröpfchen in alkalisches Bad geben (aus Ätznatron und konz. Wasserstoffperoxid) und erhitzen, abfiltrierten Niederschlag mit warmem Wasser waschen, in HCl lösen; in einem Teil der Lösung Fe nachweisen, den anderen Teil 2 mal mit 1 mL konz Salpetersäure abrauchen und mit konz. Salpetersäure und Blei(IV)-oxid aufkochen, filtrieren (Nachweisreaktion: Violettfärbung zeigt Permanganat an).
- Mangan(II) + Natronlauge + Wasserstoffperoxid ergibt Braunstein.

- Eisen(III) fällt als braunes Eisenhydroxid

Abtrennung von Aluminiumhydroxid
[Bearbeiten]Stark alkalische Lösung durch Kochen von Wasserstoffperoxid befreien (Nachweisreaktion: bei Gelbfärbung Chromat!), mit HCl neutralisieren, einige Tropfen Ammoniak und 2-3 Spatelspitzen Ammoniumchlorid zugeben, aufkochen, den weißen Aluminiumhydroxid-Niederschlag abfiltrieren und Aluminiumeinzelnachweise durchführen.
- Aluminium fällt als Aluminiumhydroxid, dass im Überschuss als farbloser Komplex gelöst wird.
![{\displaystyle \mathrm {Al^{3+}+4\ OH^{-}\longrightarrow [Al(OH)_{4}]^{-}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/10457caff6a27f33fa3b9824d39b54a9912b79af)
Abtrennung von Chrom(at) zum Nachweis von Zink
[Bearbeiten]Filtrat vom Aluminiumhydroxid-Nd. bei Gelbfärbg. mit Bariumchlorid-Lösung behandeln (nach Pufferung mit HAc/NaAc!) und Bariumchromat abfiltrieren, gelben Filterrückstand in verdünnter Schwefelsäure lösen, mit Wasserstoffperoxid und Ether schütteln (Nachweisreaktion ähnlich wie beim alkalischen Bad: zusätzl. Chromnachweis, kann bei eindeutiger Vorprobe und Gelbfärbung u.U. entfallen (Vorsicht: Etherdämpfe können sich in Flammennähe explosionsartig entzünden!). In schwach essigsaures Filtrat vom Bariumchromat Schwefelwasserstoff-Gas einleiten, weißen ZnS-Niederschlag abfiltrieren, in HCl lösen, mit NaOH kochen und - falls nötig - störende Niederschläge abfiltrieren (im Filtrat ist Zn als Hydroxidozinkat-Komplex farblos gelöst), mit HAc/NaAc puffern und erneut ZnS zum Zinkeinzelnachweis ausfällen.
- Chrom(III) wird zu einer gelben Chromatlösung oxidiert.

- Zink geht als farbloser Hydroxidozinkat-Komplex in Lösung.
![{\displaystyle \mathrm {Zn^{2+}+4\ OH^{-}\longrightarrow [Zn(OH)_{4}]^{2-}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/12c88d00e7e2ce999eb3898a1192b7f841b80e65)
Ammoniumcarbonatgruppe
[Bearbeiten]Die Gruppenfällung mit Ammoniumcarbonatlösung
[Bearbeiten]Filtrat der Ammoniumsulfidgruppe mit HCl ansäuern und Schwefelwasserstoff verkochen; aus nicht allzu verdünnter Lösung (gegf. Volumen durch Sieden auf einige mL einengen) im ammoniakalischen mit einem Überschuss an konzentrierter Ammoniumcarbonatlösung ausfällen. Bei einem pH-Wert von 8 bis 9 fallen] in (NH4)2CO3-haltiger Lösung die Carbonate von Ca(II), Sr(II) und Ba(II) mit Carbonat-Anionen. Filtrat auf Vollständigkeit der Fällung prüfen und den Niederschlag mit Ammoniumcarbonatlösung waschen.
Abtrennung des Bariums
[Bearbeiten]Carbonatniederschläge in 1-2 mL 2molarer Essigsäure (HAc) lösen und das Kohlendioxid durch Kochen vertreiben, mit 2-3 Spatelspitzen Salmiaksalz abpuffern und tropfenweise orange Dichromatlösung Cr2O72– zugeben, bis das gelbe Bariumchromat ausfällt und überstehende Lösung durch Chromate gelb gefärbt ist (Aufheben!). Niederschlag abfiltrieren, mit H2O waschen und in 2-3 Tropfen verdünnter HCl lösen. Mit etwas verdünnter oder 1 Tropfen konzentrierter Schwefelsäure das Bariumsulfat fällen und im Filter waschen (zur Prüfung der Flammenfärbung):

Untersuchung auf Strontium und Calcium
[Bearbeiten]Das Filtrat der Bariumchromatfällung wird mit 1 mL konzentrierter Ammoniumcarbonatlösung gekocht (ca. 1 min), filtriert und der Niederschlag wird chromatfrei gewaschen und danach in 5m HCl gelöst. Das Kohlendioxid wird verkocht und die Lösung geviertelt (1:2:1):
- a) ein 1. Teil wird mit Ammoniumsulfatlösung versetzt, filtriert und das Filtrat mit Ammoniumoxalatlösung auf noch vorhandene, überschüssige Calciumionen untersucht. Der Calciumoxalatniederschlag sollte eine ziegelrote Flammenfärbung ergeben.
- b) der 2. und 3. Teil werden vereinigt, mit Ammoniumoxalatlösung versetzt, um Calciumionen auszufällen (wie oben), und das Filtrat mit gesättigter Gipslösung versetzen, um das noch schwerer lösliche Strontiumsulfat auszufällen. Der Strontiumsulfatniederschlag wird mit Wasser gewaschen und auf Flammenfärbung untersucht: tiefrote Flamme (ggf. mit je einem Salzkorn Calcium- und Strontiumsalz vergleichen).
- c) der 4. Teil wird ebenfalls mit gesättigter Gipslösung versetzt, um das noch schwerer lösliche Strontiumsulfat auszufällen. Der Strontiumsulfatniederschlag wird mit Wasser gewaschen und auf Flammenfärbung untersucht: tiefrote Flamme (ggf. mit dem Niederschlag aus b) und je einem Salzkorn Calcium- und Strontiumsalz vergleichen).
| Ion | Flammen- färbung | Reaktion mit OH- | ...mit CO32- | ...mit C2O42- | ...mit CrO42- |
|---|---|---|---|---|---|
| Beryllium | keine | Be(OH)2 fällt aus | BeCO3 ist löslich | ? | BeCrO4 ist löslich |
| Magnesium | keine | Mg(OH)2 fällt aus | MgCO3 fällt aus | MgC2O4 ist löslich | MgCrO4 ist löslich |
| Calcium | ziegelrot | Ca(OH)2 fällt aus | CaCO3 fällt aus | CaC2O4 fällt aus | CaCrO4 fällt aus |
| Strontium | intensiv rot | Sr(OH)2 fällt aus | SrCO3 fällt aus | SrC2O4 ist löslich | SrCrO4 fällt aus |
| Barium | gelb-grün | Ba(OH)2 ist löslich | BaCO3 fällt aus | BaC2O4 ist löslich | BaCrO4 fällt aus |
Die Kationen der Löslichen Gruppe bleiben im Anschluss an die Salzsäure-, Schwefelwasserstoff-, Ammoniumsulfid- und Ammoniumcarbonatgruppe im Filtrat übrig. Hierzu gehören nämlich diejenigen Elemente, die mit keinem der Trennmittel schwerlösliche Niederschläge bilden: NH4+, Mg2+, K+, Na+, Li+. Diese können mittels spezifischer Nachweisreaktionen und Flammenfärbung identifiziert werden.
Da der alkalische Sturz nach der Ammoniumsulfidfällung Anfängern häufig Schwierigkeiten bereitet, kann für schwer nachweisbare Stoffe ein Kaliumhydroxidauszug aus der Ursubstanz hergestellt werden. Hierfür wird die Ursubstanz mit 3 Kaliumhydroxid-Plätzchen und 5 ml Wasser versetzt. Es fallen Kupfer, Bismut, Nickel, Cobalt, Eisen und Mangan unter Bildung schwerlöslicher Hydroxide aus. In Lösung verbleiben Antimon, Zinn, Aluminium, Zink und Chrom, die nun mit spezifischen Nachweisreaktionen nachgewiesen werden können.
Quantitative Analyse
[Bearbeiten]
Die Titrimetrie (Maßanalyse, Volumetrie) ist eine vielfach gebrauchte Methode für die Gehaltsbestimmung vor allem in wässrigen Lösungen. Das Prinzip ist sehr einfach: Man gibt mit einer Bürette Maßlösung (Titrator, Titrant, Normallösung) bekannter Konzentration zu einer Probelösung (Titrand) bis der Äquivalenzpunkt erreicht ist. Durch stöchiometrische Rechnungen kann dann die Konzentration des gesuchten Stoffes in der Probelösung bestimmt werden. Als Indikator dient entweder eine chemische Verbindung, die unter den Versuchsbedingungen am Äquivalenzpunkt die Farbe ändert, eine Messelektrode, die über ein Messgerät den Äquivalenzpunkt anzeigt (z.B. pH-Wert, Potentiometrie) oder die Verfärbung des Niederschlages (z.B. Chloridbestimmung nach Mohr).
Die Titration läßt sich in mehrere Gebiete einteilen:
- Säure-Base-Titrationen (Acidometrie, Alkalimetrie)
- Fällungsreaktion
- Komplexometrie
- Redox-Titrationen
- Spezielle Methoden wie die Zwei-Phasen-Titration nach Epton oder die Polyelektrolyttitration zur Bestimmung des kationischen Bedarfs
Als Methoden kommen die direkte Titration, bei der direkt in Probelösung titriert wird oder eine Maßlösung mit Probelösung titriert wird (inverse Titration), und die indirekte Titration, bei der entweder eine Umsetzung der Probelösung mit einer Reagenslösung bekannter Menge und Konzentration stattfindet und die Reagenslösung dann titriert wird (Rücktitration) oder der zu bestimmende Stoff nach Reagenszugabe aus dem Reagens einen Stoff freisetzt, der dann titriert wird (Substitutionstitration), in Frage.
Messgeräte
[Bearbeiten]Bei der Titration werden vor allem Volumenmessgeräte wie Büretten, Pipetten, Messzylinder und Messkolben verwendet.
-
 Bürette
Bürette -
 Peleusball auf Auslaufpipette
Peleusball auf Auslaufpipette -
 Vollpipette
Vollpipette -
 Maßkolben/Messkolben
Maßkolben/Messkolben


Ablesen
[Bearbeiten]Wasser (A) bildet durch hydrophile Wechselwirkungen mit dem Glas eine nach unten gewölbte Oberfläche (konkaver Meniskus). Bei Quecksilber (B) ist es übrigens anders herum: es kommt zu einer nach oben gewölbten Flüssigkeitsoberlfäche. Bei Wasser wird stets der untere Meniskus abgelesen, bei Stoffen, die einen konvexen Meniskus ausbilden, der obere. Meist wird der abzulesende Strich in der Skalierung durch die Lichtbrechung mit dem Wasser vergrößert und kann so bequem abgelesen werden.

Des Weiteren sollte darauf geachtet werden auf Augenhöhe abzulesen da ansonsten durch den Parallaxenfehler erhebliche Messfehler auftreten können.

Konzentrationsangaben
[Bearbeiten]Ein wichtiger Begriff in der Maßanalyse, wie auch in der gesamten quantitativen Analyse überhaupt, ist die Konzentration. Konzentrationsangaben werden in der Maßanalyse in Form der Stoffmengenkonzentration (c, Molarität, Volummolarität). Diese gibt an, wieviel Mol eines Stoffes in einem Liter Lösungsmittel gelöst sind ([c] = 1 mol/l). Anstelle der Einheit „mol/l“ wird sehr gerne die nicht-gesetzliche Einheit „Molar“ (Einheitenzeichen: M) verwendet. Streng genommen darf sie aufgrund der gesetzlichen Bestimmungen nicht verwendet werden; „M“ kann heute nur als Abkürzung von „mol/l“ verstanden werden.
Besonders in der Maßanalyse spielt auch noch die Normalität eine Rolle, die angibt, wieviele entsprechende Teilchen (z.B. Protonen) übertragen werden.
Verdünnen
[Bearbeiten]
Das Verdünnen von konzentrierten Lösungen ist in der Titrimetrie von Bedeutung, wenn die zu analysierende Substanz viel niedriger konzentriert ist als die Maßlösung. Es macht keinen Sinn, in fünf Tropfen bis zum Umschlagspunkt zu titrieren. Die Messungenauigkeit wird viel zu hoch. Dagegen kann ein Verdünnen der Maßlösung für die Bürette die Genauigkeit erhöhen, denn der Bürettenfehler (ist immer als ±0,X aufgedruckt) bleibt konstant. Der relative Fehler = Bürettenfehler/Titriervolumen wird kleiner:

Man titriert 1 mL mit einem Bürettenfehler von 0,1 mL. Egal wie gut man sich anstellt, man hat im Mittel immer 10% Abweichung. Der Wert ist so ungenau, dass das Ergebnis praktisch wertlos ist.

Titriert man dagegen 10 mL bis zum Umschlagspunkt in der gleichen Bürette, ist man gleich zehnmal so genau.

Bei 20 mL ist die Ungenauigkeit gleich nochmal halbiert.
Jedoch ist zu beachten, dass beim Verdünnen ebenfalls Ablesefehler und Gerätefehler (ebenfalls auf Auslaufpipetten und Messkolben als ±0,X gedruckt) auftreten. Der Titer oder Normalfaktor f ist ein Faktor, der die Abweichung der tatsächlichen Konzentration einer Maßlösung von der Nennkonzentration der Lösung angibt.

Daraus ergibt sich bei der Titration mit der eingestellten Lösung

Der Titer ist ein für die jeweilige Maßlösung spezifischer Wert. Je nach Bestimmungsmethode kann ein leicht unterschiedlicher Titer für ein und dieselbe Maßlösung bestimmt werden. Sinnvollerweise wird die gleiche Methode für die Messung und die Titer-Bestimmung verwendet, da die Endpunkt-Bestimmung bei jeder Methode unterschiedlich ist und so Differenzen entstehen.
Einige Maßlösungen sind so instabil, dass der Titer bei jeder Verwendung der Maßlösung neu ermittelt werden muss. Dies gilt besonders für niedrig konzentrierte Lösungen von Iod, Salzsäure, Natronlauge und Kaliumpermanganat; Iod und Chlorwasserstoff verdampfen aus der Lösung, Natriumhydroxid bildet mit dem Kohlendioxid der Luft Natriumcarbonat und Kaliumpermanganat zersetzt sich in einem autokatalytischen Prozess zu Braunstein.
Einigermaßen stabil sind höherkonzentrierte Maßlösungen von Salz- und Schwefelsäure, Natriumthiosulfat, Cer(IV), Kaliumdichromat, Silbernitrat (unter Lichtausschluss) und natürlich Urtitermaßlösungen. Aber auch diese Maßlösungen müssen mindestens einmal pro Monat geprüft werden.
Zur Bestimmung des Titers werden sog. Urtitersubstanzen verwendet. Dies sind Substanzen, die sich zum einen leicht in einer definierten Zusammensetzung (wasserfrei, bestimmtes Hydrat) herstellen lassen, ein möglichst hohes Molekulargewicht haben und sich daher leicht mit geringem Fehler abwiegen lassen. Sie sind Feststoffe wie z.B. Oxalsäure-Dihydrat und Natriumcarbonat.
Bestimmung des Titers einer Salzsäurelösung
[Bearbeiten]Als Urtitersubstanz wird Natriumcarbonat gewählt, das mit Salzsäure wie folgt reagiert:

Aus der Reaktionsgleichung ist ersichtlich, dass die halbe Stoffmenge Natriumcarbonat der verbrauchten Stoffmenge Salzsäure entspricht.
Es wird eine bestimmte Menge Natriumcarbonat, das aus einer gesättigten Lösung mit Kohlendioxid ausgefällt, gewaschen und bis zur Massenkonstanz getrocknet wurde, möglichst genau abgewogen, in Wasser gelöst und mit einem Indikator wie Methylorange versetzt. Nun wird bis zum Umschlagpunkt titriert. Aus dem Verbrauch an Maßlösung und der eingesetzten Stoffmenge an Natriumcarbonat kann die Konzentration der Salzsäurelösung bestimmt werden.
- Vorlage
- m(Na2CO3) = 0,4000 g (~3.77 mmol); c(HCl) = ca. 0,1 mol/l
- Verbrauch an Maßlösung
- 75,0 ml
- Stöchiometrie

- (gemäß Beispiel: 2 * 3,77 mmol = 7,5 mmol)



Die Salzsäurelösung hat also eine tatsächliche Konzentration von 0,1006 mol/l. Um den Titer zu errechnen, teilt man nun die gemessene tatsächliche Konzentration durch die Nennkonzentration und erhält:

Bei der Säure-Base-Titration reagieren Säuren und Basen in einer Neutralisationsreaktion miteinander. Wird eine Säure untersucht und mit einer Base titriert spricht man von Alkalimetrie. Wird eine basische Probelösung mit einer sauren Maßlösung titriert so spricht man von einer Acidimetrie.
Herstellung einer Maßlösung und Titerbestimmung
[Bearbeiten]Die Maßlösung wird in einem Messkolben hergestellt. Um den Fehler hier zu minimieren sollte ein möglichst großes Volumen angesetzt werden. Maßlösungen werden vorteilhafterweise entweder in „geraden“ Molaritäten (1 mol/l, 0,1 mol/l usw.) oder Normalitäten (0,5 mol/l entspricht 1 normal usw. beispielsweise bei Schwefelsäure) angesetzt. Als Maßlösungen kommen meist nur starke Basen und starke Säuren zum Einsatz.
Fehlerquellen
[Bearbeiten]Natron- und besonders Kalilauge verändern beim Stehen an Luft ihre Konzentration etwas. Dies geschieht aufgrund der Aufnahme von Kohlendioxid aus der Luft; dabei bildet sich das entsprechende Carbonat. Auch auf festem Natrium- und Kaliumhydroxid bildet sich leicht eine Carbonatschicht. Zur Herstellung der Lauge wird daher zunächst die entsprechende Menge Hydroxid eingewogen und dieses dann durch sehr kurzes Abspülen mit Wasser vom Carbonat befreit. Der geringe Unterschied zwischen Einwaage und tatsächlicher Masse nach dem Abspülen wird durch die Bestimmung des Titers ausgeglichen. Für sehr genaue Arbeiten ist unter Umständen das Herstellen von carbonatfreiem Wasser nötig.
Wahl der Indikatoren
[Bearbeiten]Es stehen eine große Anzahl von Indikatoren mit verschiedenen Umschlagsbereichen zur Verfügung:

Es gilt:
- Starke Säuren und starke Basen können unter Verwendung aller Indikatoren, die zwischen Methylorange und Phenolphthalein umschlagen, miteinander titriert werden.
- Schwache Säuren mit starken Laugen können nur mit Indikatoren, die im schwach alkalischen Gebiet umschlagen titriert werden (z.B. Phenolphthalein).
- Schwache Basen mit starken Säuren lassen sich nur mit Indikatoren, die im schwach sauren Bereich umschlagen titrieren (z.B. Methylorange, besser Methylrot)
- Titrationen schwacher Säuren mit schwachen Basen oder umgekehrt sind zu vermeiden, weil sie ungenaue Resultate bringen. Der richtige Indikator muss hier individuell mit Vergleichslösungen ermittelt werden.
Bestimmung der Konzentration von Salzsäure mit Natronlauge
[Bearbeiten]Zu einem bekannten Volumen an Salzsäure mit unbekannter Konzentration wird aus einer Bürette die Maßlösung ( Natronlauge mit bekannter Konzentration) bis zum Umschlagspunkt des Indikators hinzugetropft. Es handelt sich um eine Titration einer starken Säure mit einer starken Base, daher sind alle Indikatoren im Umschlagsbereich pH=4 bis pH=10 zulässig wenn im Konzentrationsbereich 0,1 mol/L mit 0,1% Bestimmungsfehler gearbeitet wird. Im Konzentrationsbereich 0,01 mol/L dürfen die pH-Grenzwerte 5 und 9 nicht unter- bzw. überschritten werden soll.
Aufgrund des Volumen der untersuchten Salzsäure und verbrauchten Menge an Natronlauge bis zum Umschlagspunkt kann unter Zuhilfenahme der Reaktionsgleichung die Konzentration der Salzsäure berechnet werden.
Die Stoffmenge n der eingesetzten Natronlauge ergibt sich aus der Stoffmengenkonzentration c und dem Volumen V

Die Reaktionsgleichung der Neutralisation lautet:
- Salzsäure und Natronlauge reagieren zu Kochsalz und Wasser

Daraus folgt: Salzsäure reagiert mit Natronlauge im Stoffmengenverhältnis eins zu eins:

Die Konzentration der Salzsäure lässt sich nun aus der bekannten Stoffmenge und dem verwendeten Volumen errechnen:

{kind=link}
Autoren
[Bearbeiten]Text
[Bearbeiten]Wikipedia
[Bearbeiten]Teile dieses Wikibooks stammen aus Wikipedia-Artikeln, die aufgrund des stark sachbuchhaften Anleitungscharakters dort unpassend waren. Die Autoren der jeweiligen Artikel sind unten separat aufgeführt. Die Listen wurden mit dem Programm ![]() Contributors von Benutzer:Duesentrieb auf dem Wikimedia Toolserver erstellt.
Contributors von Benutzer:Duesentrieb auf dem Wikimedia Toolserver erstellt.
- bis 2006-04-02 danach #Nachweis (Chemie, Methodenteil)
- 87 Wächter 2006-01-05 11:35 – 2006-03-22 17:56
- 28 82.207.144.171 (anon) 2006-01-11 12:25 – 2006-02-01 14:16
- 9 80.143.235.108 (anon) 2006-01-09 11:52 – 2006-01-09 12:05
- 2 Christoph D 2005-03-19 16:10 – 2005-10-18 18:21
- 6 80.143.89.144 (anon) 2006-01-13 16:14 – 2006-01-13 16:52
- 5 193.158.3.10 (anon) 2005-09-26 08:14 – 2005-09-26 08:27
- 4 Baultbear 2005-12-04 19:16 – 2005-12-04 19:22
- 4 80.143.110.129 (anon) 2006-01-05 11:28 – 2006-01-05 11:45
- 3 80.137.216.237 (anon) 2005-11-22 20:28 – 2005-11-22 20:40
- 3 84.166.237.225 (anon) 2005-11-23 17:11 – 2005-11-23 17:16
- 3 80.143.73.138 (anon) 2006-01-06 13:41 – 2006-01-06 13:47
- 3 80.143.244.68 (anon) 2006-01-10 12:03 – 2006-01-10 12:16
- 3 80.143.77.155 (anon) 2006-02-01 16:51 – 2006-02-01 16:53
- 2 PIGSgrame 2006-02-01 17:37 – 2006-02-03 15:14
- 2 145.254.213.29 (anon) 2004-10-24 14:49 – 2004-10-24 14:51
- 2 128.130.142.16 (anon) 2005-10-20 11:55 – 2005-10-20 11:57
- 2 Agabuga 2005-11-16 19:55 – 2005-11-16 20:02
- 2 158.64.68.1 (anon) 2005-12-13 07:40 – 2005-12-13 07:44
- 2 85.75.108.22 (anon) 2005-12-22 11:47 – 2005-12-22 11:47
- 2 Prolineserver 2006-03-06 20:01 – 2006-03-07 18:53
- 1 Hati 2006-03-11 15:54 – 2006-03-11 16:41
- 2 88.73.214.237 (anon) 2006-03-19 19:42 – 2006-03-19 19:45
- 1 ThomasM 2004-03-14 18:39 – 2004-03-14 18:39
- 1 HenHei 2004-03-14 18:51 – 2004-03-14 18:51
- 1 Rec 2004-03-20 12:08 – 2004-03-20 12:08
- 1 80.134.250.78 (anon) 2004-12-07 09:07 – 2004-12-07 09:07
- 1 62.226.215.189 (anon) 2005-03-02 16:36 – 2005-03-02 16:36
- 1 212.204.24.73 (anon) 2005-03-26 12:39 – 2005-03-26 12:39
- 1 213.54.225.30 (anon) 2005-07-23 16:35 – 2005-07-23 16:35
- 1 84.175.127.175 (anon) 2005-10-18 12:46 – 2005-10-18 12:46
- 1 84.189.120.107 (anon) 2005-11-21 22:07 – 2005-11-21 22:07
- 1 213.39.153.228 (anon) 2005-11-30 12:23 – 2005-11-30 12:23
- 1 80.143.81.126 (anon) 2006-01-05 08:01 – 2006-01-05 08:01
- 1 86.56.9.11 (anon) 2006-01-08 17:32 – 2006-01-08 17:32
- 1 85.74.61.20 (anon) 2006-01-12 21:24 – 2006-01-12 21:24
- 1 Elya 2006-01-12 21:38 – 2006-01-12 21:38
- 1 80.171.73.187 (anon) 2006-01-13 19:40 – 2006-01-13 19:40
- 1 80.143.75.169 (anon) 2006-01-17 17:43 – 2006-01-17 17:43
- 1 84.180.43.201 (anon) 2006-01-20 17:42 – 2006-01-20 17:42
- 1 84.139.27.1 (anon) 2006-01-25 17:40 – 2006-01-25 17:40
- 1 80.184.171.204 (anon) 2006-01-26 12:34 – 2006-01-26 12:34
- 1 62.203.4.203 (anon) 2006-01-28 09:36 – 2006-01-28 09:36
- 1 80.133.104.149 (anon) 2006-01-29 17:31 – 2006-01-29 17:31
- 1 Cottbus 2006-02-16 14:21 – 2006-02-16 14:21
- 1 Mkill 2006-02-17 01:16 – 2006-02-17 01:16
- 1 132.195.66.162 (anon) 2006-02-23 07:45 – 2006-02-23 07:45
- 1 87.122.146.57 (anon) 2006-03-04 09:21 – 2006-03-04 09:21
- 1 83.135.218.4 (anon) 2006-03-06 18:52 – 2006-03-06 18:52
- 1 80.143.94.149 (anon) 2006-03-11 12:08 – 2006-03-11 12:08
- 1 193.171.131.240 (anon) 2006-03-18 12:54 – 2006-03-18 12:54
- 1 Wg0867 2006-03-25 23:48 – 2006-03-25 23:48
- 1 89.51.248.40 (anon) 2006-03-30 18:26 – 2006-03-30 18:26
- bis 2006-11-26 danach Inhalt ausgelagert nach w:Nachweis (Chemie), w:Nachweise für Anionen, w:Nachweise für Kationen, w:Nachweise organischer Stoffe; siehe w:Wikipedia:Redaktion Chemie/Archiv/2006/Dezember#Kationennachweise
- 8 84.151.235.36 (anon) 2006-10-09 10:28 – 2006-10-09 10:35
- 3 NEUROtiker 2006-09-17 20:55 – 2006-11-26 23:34
- 4 84.170.137.15 (anon) 2006-05-21 19:16 – 2006-05-21 19:20
- 1 Stefan Horn 2006-07-27 07:43 – 2006-07-27 07:47
- 1 Wg0867 2006-04-02 21:12 – 2006-04-02 21:36
- 2 84.170.8.76 (anon) 2006-06-20 14:27 – 2006-06-20 14:28
- 2 W!B: 2006-08-02 01:24 – 2006-08-06 16:47
- 2 217.230.58.106 (anon) 2006-10-18 14:55 – 2006-10-18 14:56
- 1 Hangy 2006-04-26 16:27 – 2006-04-26 16:27
- 1 84.136.219.94 (anon) 2006-05-10 16:54 – 2006-05-10 16:54
- 1 84.180.147.186 (anon) 2006-06-12 17:38 – 2006-06-12 17:38
- 1 80.132.108.240 (anon) 2006-07-27 11:15 – 2006-07-27 11:15
- 1 Mkill 2006-07-29 20:08 – 2006-07-29 20:08
- 1 84.136.206.55 (anon) 2006-07-30 15:58 – 2006-07-30 15:58
- 1 Onkel Markus 2006-08-21 15:22 – 2006-08-21 15:22
- 1 217.255.172.186 (anon) 2006-08-24 08:22 – 2006-08-24 08:22
- 1 WAH 2006-08-24 13:38 – 2006-08-24 13:38
- 1 87.78.157.232 (anon) 2006-09-14 14:15 – 2006-09-14 14:15
- 1 62.178.201.41 (anon) 2006-09-15 20:39 – 2006-09-15 20:39
- 1 88.64.190.51 (anon) 2006-10-25 15:23 – 2006-10-25 15:23
- 1 217.227.215.186 (anon) 2006-11-12 12:45 – 2006-11-12 12:45
- 1 87.168.222.228 (anon) 2006-11-22 05:05 – 2006-11-22 05:05
- 41 Siegert 2007-03-04 07:16 – 2008-08-10 17:02
- 18 Kuebi 2008-01-25 08:55 – 2008-01-25 09:02
- 4 NEUROtiker 2006-11-26 22:23 – 2008-04-21 17:39
- 7 217.93.173.69 (anon) 2006-12-14 14:24 – 2006-12-14 15:13
- 3 84.173.201.201 (anon) 2007-03-01 21:37 – 2007-03-01 21:40
- 3 90.186.37.30 (anon) 2007-04-28 15:49 – 2007-04-28 15:51
- 3 90.186.25.28 (anon) 2007-04-30 14:42 – 2007-04-30 14:43
- 3 141.43.142.19 (anon) 2008-01-11 17:58 – 2008-01-11 18:25
- 3 Matthias M. 2008-02-26 23:27 – 2008-05-18 19:38
- 1 Hystrix 2007-06-26 19:58 – 2007-10-23 12:12
- 2 Jpidtfaz 2008-07-23 18:32 – 2008-07-23 18:32
- 1 195.93.60.66 (anon) 2007-01-05 15:18 – 2007-01-05 15:18
- 1 139.14.30.170 (anon) 2007-01-26 13:13 – 2007-01-26 13:13
- 1 141.52.232.84 (anon) 2007-02-02 09:06 – 2007-02-02 09:06
- 1 84.150.89.86 (anon) 2007-02-25 14:28 – 2007-02-25 14:28
- 1 84.185.221.242 (anon) 2007-04-11 12:40 – 2007-04-11 12:40
- 1 82.82.189.32 (anon) 2007-05-19 15:28 – 2007-05-19 15:28
- 1 84.178.253.236 (anon) 2007-09-08 17:54 – 2007-09-08 17:54
- 1 89.49.225.230 (anon) 2007-10-06 14:25 – 2007-10-06 14:25
- 1 80.63.151.42 (anon) 2007-10-23 09:34 – 2007-10-23 09:34
- 1 87.162.81.70 (anon) 2007-11-15 22:10 – 2007-11-15 22:10
- 1 84.58.36.135 (anon) 2008-02-06 22:29 – 2008-02-06 22:29
- 1 77.5.242.50 (anon) 2008-03-15 15:59 – 2008-03-15 15:59
- 1 62.47.7.104 (anon) 2008-04-04 09:45 – 2008-04-04 09:45
- 1 Muck31 2008-04-06 12:47 – 2008-04-06 12:47
- 1 129.217.132.31 (anon) 2008-04-21 13:55 – 2008-04-21 13:55
- 1 Johnny Controletti 2008-04-21 14:20 – 2008-04-21 14:20
- 1 89.247.229.20 (anon) 2008-06-01 10:43 – 2008-06-01 10:43
- 1 79.196.247.141 (anon) 2008-09-08 13:16 – 2008-09-08 13:16
- 1 84.186.113.38 (anon) 2008-11-11 12:35 – 2008-11-11 12:35
- 1 Leyo 2008-12-06 21:28 – 2008-12-06 21:28
- 71 Siegert 2007-03-16 18:27 – 2008-07-06 09:35
- 1 Wächter 2006-09-08 11:47 – 2008-04-04 10:30
- 7 91.1.226.160 (anon) 2007-06-04 17:41 – 2007-06-04 18:01
- 2 NEUROtiker 2006-11-26 22:16 – 2007-01-05 22:15
- 4 87.122.15.149 (anon) 2007-02-13 07:22 – 2007-02-13 07:23
- 2 217.235.74.111 (anon) 2007-03-12 20:36 – 2007-03-12 20:36
- 2 Matthias M. 2008-02-26 23:10 – 2008-05-18 19:37
- 1 88.70.118.175 (anon) 2006-11-15 22:11 – 2006-11-15 22:11
- 1 Tango8 2006-12-12 23:02 – 2006-12-12 23:02
- 1 80.129.14.97 (anon) 2006-12-15 00:01 – 2006-12-15 00:01
- 1 89.51.124.18 (anon) 2007-01-05 20:44 – 2007-01-05 20:44
- 1 139.14.30.170 (anon) 2007-01-26 13:13 – 2007-01-26 13:13
- 1 Akkarin 2007-02-13 07:24 – 2007-02-13 07:24
- 1 84.173.234.212 (anon) 2007-02-21 23:24 – 2007-02-21 23:24
- 1 Blaufisch 2007-03-17 09:16 – 2007-03-17 09:16
- 1 195.93.60.100 (anon) 2007-03-22 18:05 – 2007-03-22 18:05
- 1 81.189.67.108 (anon) 2007-04-10 15:10 – 2007-04-10 15:10
- 1 84.185.204.10 (anon) 2007-05-09 15:41 – 2007-05-09 15:41
- 1 212.183.65.13 (anon) 2007-05-27 15:05 – 2007-05-27 15:05
- 1 Tafkas 2007-06-04 17:56 – 2007-06-04 17:56
- 1 217.228.105.26 (anon) 2007-06-08 20:06 – 2007-06-08 20:06
- 1 84.178.65.250 (anon) 2007-07-19 18:17 – 2007-07-19 18:17
- 1 130.133.10.10 (anon) 2007-08-26 14:03 – 2007-08-26 14:03
- 1 Muck31 2007-10-14 21:08 – 2007-10-14 21:08
- 1 88.73.38.51 (anon) 2007-10-30 21:58 – 2007-10-30 21:58
- 1 82.135.87.215 (anon) 2008-01-10 19:06 – 2008-01-10 19:06
- 1 Don Magnifico 2008-01-27 11:48 – 2008-01-27 11:48
- 1 141.30.211.71 (anon) 2008-03-11 00:52 – 2008-03-11 00:52
- 1 84.179.221.32 (anon) 2008-04-30 09:59 – 2008-04-30 09:59
- 1 FK1954 2008-05-25 15:47 – 2008-05-25 15:47
- 1 79.194.62.11 (anon) 2008-11-11 07:26 – 2008-11-11 07:26
- 1 HaSee 2009-01-23 13:00 – 2009-01-23 13:00
- 7 Wächter 2006-01-21 08:18 – 2008-03-03 07:35
- 2 Matthias M. 2008-03-04 08:33 – 2008-03-12 16:33
- 2 HolgerB 2006-01-23 17:45 – 2007-02-10 18:23
- 2 62.203.4.203 (anon) 2006-01-28 13:26 – 2006-01-28 13:28
- 1 84.168.241.111 (anon) 2006-04-17 14:51 – 2006-04-17 14:51
- 1 Ephraim33 2006-07-29 17:48 – 2006-07-29 17:48
- 1 139.14.30.170 (anon) 2007-01-26 13:16 – 2007-01-26 13:16
- 1 62.227.119.38 (anon) 2007-05-10 22:24 – 2007-05-10 22:24
- 20 Wächter 2006-01-21 09:22 – 2006-03-11 14:52
- 1 FrankOE 2006-02-22 10:36 – 2006-03-02 10:04
- 2 Matthias M. 2008-02-27 12:56 – 2008-03-08 16:48
- 3 80.143.78.175 (anon) 2006-02-13 17:33 – 2006-02-13 17:38
- 2 213.39.176.29 (anon) 2007-04-26 11:28 – 2007-04-26 11:28
- 2 88.207.212.42 (anon) 2008-03-03 14:22 – 2008-03-03 14:26
- 1 84.156.207.165 (anon) 2006-05-16 16:52 – 2006-05-16 16:52
- 1 139.14.30.170 (anon) 2007-01-26 13:13 – 2007-01-26 13:13
- 1 84.56.232.58 (anon) 2007-01-26 15:04 – 2007-01-26 15:04
- 1 128.176.223.173 (anon) 2007-02-22 12:14 – 2007-02-22 12:14
- 1 62.227.119.38 (anon) 2007-05-10 22:08 – 2007-05-10 22:08
- 1 87.180.7.137 (anon) 2007-05-29 16:20 – 2007-05-29 16:20
- 1 87.180.9.92 (anon) 2007-06-05 17:41 – 2007-06-05 17:41
- 1 193.171.244.138 (anon) 2008-01-08 13:38 – 2008-01-08 13:38
- 1 Don Magnifico 2008-01-10 13:16 – 2008-01-10 13:16
- 14 Wächter 2006-01-21 15:39 – 2008-04-18 10:53
- 2 Matthias M. 2008-03-04 08:37 – 2008-03-08 15:37
- 1 84.190.247.89 (anon) 2006-02-05 18:07 – 2006-02-05 18:07
- 1 Benjaminw 2006-05-01 10:56 – 2006-05-01 10:56
- 1 84.187.21.167 (anon) 2006-05-11 09:17 – 2006-05-11 09:17
- 1 AHZ 2006-05-26 13:06 – 2006-05-26 13:06
- 1 139.14.30.170 (anon) 2007-01-26 13:14 – 2007-01-26 13:14
- 1 128.176.223.172 (anon) 2007-03-22 07:38 – 2007-03-22 07:38
- 1 88.64.123.194 (anon) 2007-11-04 19:17 – 2007-11-04 19:17
- 4 Wächter 2006-01-22 13:47 – 2006-03-12 10:55
- 2 Matthias M. 2008-03-04 08:38 – 2008-03-08 16:08
- 1 80.143.77.155 (anon) 2006-02-01 16:48 – 2006-02-01 16:48
- 1 84.187.224.171 (anon) 2006-04-21 16:39 – 2006-04-21 16:39
- 1 84.58.169.132 (anon) 2006-05-07 20:16 – 2006-05-07 20:16
- 1 129.13.72.33 (anon) 2006-09-25 20:12 – 2006-09-25 20:12
- 1 139.14.30.170 (anon) 2007-01-26 13:15 – 2007-01-26 13:15
- 1 193.18.239.4 (anon) 2007-05-28 09:25 – 2007-05-28 09:25
- 5 Wächter 2006-01-22 14:19 – 2006-03-12 10:49
- 2 Matthias M. 2008-03-04 08:43 – 2008-03-08 16:21
- 1 139.14.30.170 (anon) 2007-01-26 13:11 – 2007-01-26 13:11
- 1 89.245.14.251 (anon) 2007-09-24 17:39 – 2007-09-24 17:39
- 1 Milky0208 2008-02-14 16:08 – 2008-02-14 16:08
- 6 RolfS 2004-04-10 17:03 – 2004-11-25 18:21
- 5 Roland.chem 2007-01-21 13:36 – 2009-02-08 13:18
- 2 Flokru 2004-05-10 22:30 – 2004-05-10 22:35
- 1 Solid State 2007-01-17 21:36 – 2007-06-24 18:11
- 3 WikiAutor 2006-01-14 11:10 – 2006-01-21 19:29
- 3 84.135.202.125 (anon) 2006-03-17 07:41 – 2006-03-17 07:43
- 3 82.212.52.181 (anon) 2006-12-12 16:50 – 2006-12-12 16:51
- 2 82.212.60.141 (anon) 2005-02-05 22:35 – 2005-02-05 22:47
- 2 172.182.152.116 (anon) 2006-02-01 17:44 – 2006-02-01 17:45
- 2 87.79.138.148 (anon) 2007-07-29 19:15 – 2007-07-29 19:16
- 2 84.168.90.2 (anon) 2006-03-15 18:31 – 2006-03-15 18:42
- 1 BirgitLachner 2004-04-09 20:09 – 2004-04-09 20:12
- 2 83.99.65.186 (anon) 2007-06-24 14:45 – 2007-06-24 14:46
- 2 84.156.64.212 (anon) 2005-06-06 15:02 – 2005-06-06 15:02
- 2 80.139.107.167 (anon) 2007-05-02 14:38 – 2007-05-02 14:39
- 1 87.122.55.30 (anon) 2005-11-28 22:27 – 2005-11-28 22:27
- 1 217.93.254.204 (anon) 2008-09-03 17:26 – 2008-09-03 17:26
- 1 84.173.162.156 (anon) 2006-05-31 14:10 – 2006-05-31 14:10
- 1 84.132.108.19 (anon) 2007-05-10 06:17 – 2007-05-10 06:17
- 1 213.3.249.224 (anon) 2007-01-22 21:10 – 2007-01-22 21:10
- 1 217.85.230.216 (anon) 2006-06-21 14:04 – 2006-06-21 14:04
- 1 217.228.156.108 (anon) 2007-02-04 15:44 – 2007-02-04 15:44
- 1 93.129.219.64 (anon) 2009-01-20 15:28 – 2009-01-20 15:28
- 1 84.58.108.5 (anon) 2006-07-26 17:53 – 2006-07-26 17:53
- 1 145.254.239.130 (anon) 2005-02-13 14:18 – 2005-02-13 14:18
- 1 134.147.110.105 (anon) 2007-02-20 15:27 – 2007-02-20 15:27
- 1 212.152.169.204 (anon) 2009-02-10 14:38 – 2009-02-10 14:38
- 1 84.169.127.146 (anon) 2006-09-17 11:48 – 2006-09-17 11:48
- 1 91.89.9.41 (anon) 2009-03-03 13:01 – 2009-03-03 13:01
- 1 84.135.231.107 (anon) 2005-06-13 06:59 – 2005-06-13 06:59
- 1 69.152.221.194 (anon) 2006-12-06 13:10 – 2006-12-06 13:10
- 1 Terabyte 2005-06-13 19:07 – 2005-06-13 19:07
- 1 89.54.72.56 (anon) 2006-03-20 13:58 – 2006-03-20 13:58
- 1 217.88.44.157 (anon) 2007-04-13 10:10 – 2007-04-13 10:10
- 1 143.107.55.161 (anon) 2006-05-13 00:16 – 2006-05-13 00:16
- 1 85.176.3.196 (anon) 2007-04-17 22:02 – 2007-04-17 22:02
- 1 217.82.176.166 (anon) 2004-06-02 15:09 – 2004-06-02 15:09
- 1 81.172.157.23 (anon) 2007-01-08 18:27 – 2007-01-08 18:27
- 1 80.134.234.98 (anon) 2006-05-30 13:04 – 2006-05-30 13:04
- 1 Louis Bafrance 2007-05-02 14:53 – 2007-05-02 14:53
- 1 Fedi 2004-07-13 18:31 – 2004-07-13 18:31
- 1 217.186.183.28 (anon) 2005-12-12 20:54 – 2005-12-12 20:54
- 1 Hoffmeier 2006-06-01 03:49 – 2006-06-01 03:49
- 1 217.251.104.113 (anon) 2004-12-08 18:44 – 2004-12-08 18:44
- 1 Talos 2006-01-19 21:15 – 2006-01-19 21:15
- 1 217.88.222.227 (anon) 2006-06-28 13:54 – 2006-06-28 13:54
- 1 Sumpfschnecke 2009-02-06 15:54 – 2009-02-06 15:54
- 1 131.246.90.63 (anon) 2007-02-26 13:17 – 2007-02-26 13:17
- 1 89.13.186.88 (anon) 2006-11-15 17:58 – 2006-11-15 17:58
- 1 Dr.cueppers 2007-11-02 15:10 – 2007-11-02 15:10
- 1 87.139.52.245 (anon) 2007-03-20 09:27 – 2007-03-20 09:27
- 1 88.70.112.55 (anon) 2006-11-19 22:04 – 2006-11-19 22:04
- 1 85.180.26.200 (anon) 2008-04-25 07:16 – 2008-04-25 07:16
- 1 134.99.18.167 (anon) 2005-06-13 19:05 – 2005-06-13 19:05
- 1 62.47.144.132 (anon) 2006-03-18 18:24 – 2006-03-18 18:24
- 1 132.199.38.123 (anon) 2006-04-20 13:34 – 2006-04-20 13:34
- 1 80.145.33.113 (anon) 2004-05-27 13:30 – 2004-05-27 13:30
- 1 80.108.228.220 (anon) 2007-01-08 17:29 – 2007-01-08 17:29
- 1 84.155.222.140 (anon) 2005-10-26 20:43 – 2005-10-26 20:43
Bilder
[Bearbeiten]- Datei:Boratflamme.jpg - GNU FDL - Benutzer:Ertua
- Datei:Leuchtprobe1.jpg, Bild:Leuchtprobe2.jpg - GNU FDL -
 User:The viewer (David Mülheims)
User:The viewer (David Mülheims) - Datei:Sulfidniederschläge.jpg - GNU FDL - Benutzer:Wächter
- Datei:Reading the meniscus.png - GNU FDL - User:Jleedev
{kind=link}
{kind=link}
Lizenzbestimmungen
[Bearbeiten]Creative Commons Attribution Share-Alike 3.0 Unported
[Bearbeiten]Deed
[Bearbeiten]Diese "Commons Deed" ist lediglich eine vereinfachte Zusammenfassung des rechtsverbindlichen Lizenzvertrages (Abschnitt "License") in allgemeinverständlicher Sprache.
Sie dürfen:
- das Werk bzw. den Inhalt vervielfältigen, verbreiten und öffentlich zugänglich machen
- Abwandlungen und Bearbeitungen des Werkes bzw. Inhaltes anfertigen
Zu den folgenden Bedingungen:


- Namensnennung — Sie müssen den Namen des Autors/Rechteinhabers in der von ihm festgelegten Weise nennen.
- Weitergabe unter gleichen Bedingungen — Wenn Sie das lizenzierte Werk bzw. den lizenzierten Inhalt bearbeiten, abwandeln oder in anderer Weise erkennbar als Grundlage für eigenes Schaffen verwenden, dürfen Sie die daraufhin neu entstandenen Werke bzw. Inhalte nur unter Verwendung von Lizenzbedingungen weitergeben, die mit denen dieses Lizenzvertrages identisch, vergleichbar oder kompatibel sind.
Wobei gilt:
- Verzichtserklärung — Jede der vorgenannten Bedingungen kann aufgehoben werden, sofern Sie die ausdrückliche Einwilligung des Rechteinhabers dazu erhalten.
- Sonstige Rechte — Die Lizenz hat keinerlei Einfluss auf die folgenden Rechte:
- Die gesetzlichen Schranken des Urheberrechts und sonstigen Befugnisse zur privaten Nutzung;
- Das Urheberpersönlichkeitsrecht des Rechteinhabers;
- Rechte anderer Personen, entweder am Lizenzgegenstand selber oder bezüglich seiner Verwendung, zum Beispiel Persönlichkeitsrechte abgebildeter Personen.
- Hinweis — Im Falle einer Verbreitung müssen Sie anderen alle Lizenzbedingungen mitteilen, die für dieses Werk gelten. Am einfachsten ist es, an entsprechender Stelle einen Link auf http://creativecommons.org/licenses/by-sa/3.0/deed.de einzubinden.
Haftungsbeschränkung
Die „Commons Deed“ ist kein Lizenzvertrag. Sie ist lediglich ein Referenztext, der den zugrundeliegenden Lizenzvertrag übersichtlich und in allgemeinverständlicher Sprache aber auch stark vereinfacht wiedergibt. Die Deed selbst entfaltet keine juristische Wirkung und erscheint im eigentlichen Lizenzvertrag nicht.
License
[Bearbeiten]CREATIVE COMMONS CORPORATION IS NOT A LAW FIRM AND DOES NOT PROVIDE LEGAL SERVICES. DISTRIBUTION OF THIS LICENSE DOES NOT CREATE AN ATTORNEY-CLIENT RELATIONSHIP. CREATIVE COMMONS PROVIDES THIS INFORMATION ON AN "AS-IS" BASIS. CREATIVE COMMONS MAKES NO WARRANTIES REGARDING THE INFORMATION PROVIDED, AND DISCLAIMS LIABILITY FOR DAMAGES RESULTING FROM ITS USE.}}
THE WORK (AS DEFINED BELOW) IS PROVIDED UNDER THE TERMS OF THIS CREATIVE COMMONS PUBLIC LICENSE ("CCPL" OR "LICENSE"). THE WORK IS PROTECTED BY COPYRIGHT AND/OR OTHER APPLICABLE LAW. ANY USE OF THE WORK OTHER THAN AS AUTHORIZED UNDER THIS LICENSE OR COPYRIGHT LAW IS PROHIBITED.
BY EXERCISING ANY RIGHTS TO THE WORK PROVIDED HERE, YOU ACCEPT AND AGREE TO BE BOUND BY THE TERMS OF THIS LICENSE. TO THE EXTENT THIS LICENSE MAY BE CONSIDERED TO BE A CONTRACT, THE LICENSOR GRANTS YOU THE RIGHTS CONTAINED HERE IN CONSIDERATION OF YOUR ACCEPTANCE OF SUCH TERMS AND CONDITIONS.
1. Definitions
[Bearbeiten]- "Adaptation" means a work based upon the Work, or upon the Work and other pre-existing works, such as a translation, adaptation, derivative work, arrangement of music or other alterations of a literary or artistic work, or phonogram or performance and includes cinematographic adaptations or any other form in which the Work may be recast, transformed, or adapted including in any form recognizably derived from the original, except that a work that constitutes a Collection will not be considered an Adaptation for the purpose of this License. For the avoidance of doubt, where the Work is a musical work, performance or phonogram, the synchronization of the Work in timed-relation with a moving image ("synching") will be considered an Adaptation for the purpose of this License.
- "Collection" means a collection of literary or artistic works, such as encyclopedias and anthologies, or performances, phonograms or broadcasts, or other works or subject matter other than works listed in Section 1(f) below, which, by reason of the selection and arrangement of their contents, constitute intellectual creations, in which the Work is included in its entirety in unmodified form along with one or more other contributions, each constituting separate and independent works in themselves, which together are assembled into a collective whole. A work that constitutes a Collection will not be considered an Adaptation (as defined below) for the purposes of this License.
- "Creative Commons Compatible License" means a license that is listed at http://creativecommons.org/compatiblelicenses that has been approved by Creative Commons as being essentially equivalent to this License, including, at a minimum, because that license: (i) contains terms that have the same purpose, meaning and effect as the License Elements of this License; and, (ii) explicitly permits the relicensing of adaptations of works made available under that license under this License or a Creative Commons jurisdiction license with the same License Elements as this License.
- "Distribute" means to make available to the public the original and copies of the Work or Adaptation, as appropriate, through sale or other transfer of ownership.
- "License Elements" means the following high-level license attributes as selected by Licensor and indicated in the title of this License: Attribution, ShareAlike.
- "Licensor" means the individual, individuals, entity or entities that offer(s) the Work under the terms of this License.
- "Original Author" means, in the case of a literary or artistic work, the individual, individuals, entity or entities who created the Work or if no individual or entity can be identified, the publisher; and in addition (i) in the case of a performance the actors, singers, musicians, dancers, and other persons who act, sing, deliver, declaim, play in, interpret or otherwise perform literary or artistic works or expressions of folklore; (ii) in the case of a phonogram the producer being the person or legal entity who first fixes the sounds of a performance or other sounds; and, (iii) in the case of broadcasts, the organization that transmits the broadcast.
- "Work" means the literary and/or artistic work offered under the terms of this License including without limitation any production in the literary, scientific and artistic domain, whatever may be the mode or form of its expression including digital form, such as a book, pamphlet and other writing; a lecture, address, sermon or other work of the same nature; a dramatic or dramatico-musical work; a choreographic work or entertainment in dumb show; a musical composition with or without words; a cinematographic work to which are assimilated works expressed by a process analogous to cinematography; a work of drawing, painting, architecture, sculpture, engraving or lithography; a photographic work to which are assimilated works expressed by a process analogous to photography; a work of applied art; an illustration, map, plan, sketch or three-dimensional work relative to geography, topography, architecture or science; a performance; a broadcast; a phonogram; a compilation of data to the extent it is protected as a copyrightable work; or a work performed by a variety or circus performer to the extent it is not otherwise considered a literary or artistic work.
- "You" means an individual or entity exercising rights under this License who has not previously violated the terms of this License with respect to the Work, or who has received express permission from the Licensor to exercise rights under this License despite a previous violation.
- "Publicly Perform" means to perform public recitations of the Work and to communicate to the public those public recitations, by any means or process, including by wire or wireless means or public digital performances; to make available to the public Works in such a way that members of the public may access these Works from a place and at a place individually chosen by them; to perform the Work to the public by any means or process and the communication to the public of the performances of the Work, including by public digital performance; to broadcast and rebroadcast the Work by any means including signs, sounds or images.
- "Reproduce" means to make copies of the Work by any means including without limitation by sound or visual recordings and the right of fixation and reproducing fixations of the Work, including storage of a protected performance or phonogram in digital form or other electronic medium.
2. Fair Dealing Rights
[Bearbeiten]Nothing in this License is intended to reduce, limit, or restrict any uses free from copyright or rights arising from limitations or exceptions that are provided for in connection with the copyright protection under copyright law or other applicable laws.
3. License Grant
[Bearbeiten]Subject to the terms and conditions of this License, Licensor hereby grants You a worldwide, royalty-free, non-exclusive, perpetual (for the duration of the applicable copyright) license to exercise the rights in the Work as stated below:
- to Reproduce the Work, to incorporate the Work into one or more Collections, and to Reproduce the Work as incorporated in the Collections;
- to create and Reproduce Adaptations provided that any such Adaptation, including any translation in any medium, takes reasonable steps to clearly label, demarcate or otherwise identify that changes were made to the original Work. For example, a translation could be marked "The original work was translated from English to Spanish," or a modification could indicate "The original work has been modified.";
- to Distribute and Publicly Perform the Work including as incorporated in Collections; and,
- to Distribute and Publicly Perform Adaptations.
- For the avoidance of doubt:
- Non-waivable Compulsory License Schemes. In those jurisdictions in which the right to collect royalties through any statutory or compulsory licensing scheme cannot be waived, the Licensor reserves the exclusive right to collect such royalties for any exercise by You of the rights granted under this License;
- Waivable Compulsory License Schemes. In those jurisdictions in which the right to collect royalties through any statutory or compulsory licensing scheme can be waived, the Licensor waives the exclusive right to collect such royalties for any exercise by You of the rights granted under this License; and,
- Voluntary License Schemes. The Licensor waives the right to collect royalties, whether individually or, in the event that the Licensor is a member of a collecting society that administers voluntary licensing schemes, via that society, from any exercise by You of the rights granted under this License.
The above rights may be exercised in all media and formats whether now known or hereafter devised. The above rights include the right to make such modifications as are technically necessary to exercise the rights in other media and formats. Subject to Section 8(f), all rights not expressly granted by Licensor are hereby reserved.
4. Restrictions
[Bearbeiten]The license granted in Section 3 above is expressly made subject to and limited by the following restrictions:
- You may Distribute or Publicly Perform the Work only under the terms of this License. You must include a copy of, or the Uniform Resource Identifier (URI) for, this License with every copy of the Work You Distribute or Publicly Perform. You may not offer or impose any terms on the Work that restrict the terms of this License or the ability of the recipient of the Work to exercise the rights granted to that recipient under the terms of the License. You may not sublicense the Work. You must keep intact all notices that refer to this License and to the disclaimer of warranties with every copy of the Work You Distribute or Publicly Perform. When You Distribute or Publicly Perform the Work, You may not impose any effective technological measures on the Work that restrict the ability of a recipient of the Work from You to exercise the rights granted to that recipient under the terms of the License. This Section 4(a) applies to the Work as incorporated in a Collection, but this does not require the Collection apart from the Work itself to be made subject to the terms of this License. If You create a Collection, upon notice from any Licensor You must, to the extent practicable, remove from the Collection any credit as required by Section 4(c), as requested. If You create an Adaptation, upon notice from any Licensor You must, to the extent practicable, remove from the Adaptation any credit as required by Section 4(c), as requested.
- You may Distribute or Publicly Perform an Adaptation only under the terms of: (i) this License; (ii) a later version of this License with the same License Elements as this License; (iii) a Creative Commons jurisdiction license (either this or a later license version) that contains the same License Elements as this License (e.g., Attribution-ShareAlike 3.0 US)); (iv) a Creative Commons Compatible License. If you license the Adaptation under one of the licenses mentioned in (iv), you must comply with the terms of that license. If you license the Adaptation under the terms of any of the licenses mentioned in (i), (ii) or (iii) (the "Applicable License"), you must comply with the terms of the Applicable License generally and the following provisions: (I) You must include a copy of, or the URI for, the Applicable License with every copy of each Adaptation You Distribute or Publicly Perform; (II) You may not offer or impose any terms on the Adaptation that restrict the terms of the Applicable License or the ability of the recipient of the Adaptation to exercise the rights granted to that recipient under the terms of the Applicable License; (III) You must keep intact all notices that refer to the Applicable License and to the disclaimer of warranties with every copy of the Work as included in the Adaptation You Distribute or Publicly Perform; (IV) when You Distribute or Publicly Perform the Adaptation, You may not impose any effective technological measures on the Adaptation that restrict the ability of a recipient of the Adaptation from You to exercise the rights granted to that recipient under the terms of the Applicable License. This Section 4(b) applies to the Adaptation as incorporated in a Collection, but this does not require the Collection apart from the Adaptation itself to be made subject to the terms of the Applicable License.
- If You Distribute, or Publicly Perform the Work or any Adaptations or Collections, You must, unless a request has been made pursuant to Section 4(a), keep intact all copyright notices for the Work and provide, reasonable to the medium or means You are utilizing: (i) the name of the Original Author (or pseudonym, if applicable) if supplied, and/or if the Original Author and/or Licensor designate another party or parties (e.g., a sponsor institute, publishing entity, journal) for attribution ("Attribution Parties") in Licensor's copyright notice, terms of service or by other reasonable means, the name of such party or parties; (ii) the title of the Work if supplied; (iii) to the extent reasonably practicable, the URI, if any, that Licensor specifies to be associated with the Work, unless such URI does not refer to the copyright notice or licensing information for the Work; and (iv) , consistent with Section 3(b), in the case of an Adaptation, a credit identifying the use of the Work in the Adaptation (e.g., "French translation of the Work by Original Author," or "Screenplay based on original Work by Original Author"). The credit required by this Section 4(c) may be implemented in any reasonable manner; provided, however, that in the case of a Adaptation or Collection, at a minimum such credit will appear, if a credit for all contributing authors of the Adaptation or Collection appears, then as part of these credits and in a manner at least as prominent as the credits for the other contributing authors. For the avoidance of doubt, You may only use the credit required by this Section for the purpose of attribution in the manner set out above and, by exercising Your rights under this License, You may not implicitly or explicitly assert or imply any connection with, sponsorship or endorsement by the Original Author, Licensor and/or Attribution Parties, as appropriate, of You or Your use of the Work, without the separate, express prior written permission of the Original Author, Licensor and/or Attribution Parties.
- Except as otherwise agreed in writing by the Licensor or as may be otherwise permitted by applicable law, if You Reproduce, Distribute or Publicly Perform the Work either by itself or as part of any Adaptations or Collections, You must not distort, mutilate, modify or take other derogatory action in relation to the Work which would be prejudicial to the Original Author's honor or reputation. Licensor agrees that in those jurisdictions (e.g. Japan), in which any exercise of the right granted in Section 3(b) of this License (the right to make Adaptations) would be deemed to be a distortion, mutilation, modification or other derogatory action prejudicial to the Original Author's honor and reputation, the Licensor will waive or not assert, as appropriate, this Section, to the fullest extent permitted by the applicable national law, to enable You to reasonably exercise Your right under Section 3(b) of this License (right to make Adaptations) but not otherwise.
5. Representations, Warranties and Disclaimer
[Bearbeiten]UNLESS OTHERWISE MUTUALLY AGREED TO BY THE PARTIES IN WRITING, LICENSOR OFFERS THE WORK AS-IS AND MAKES NO REPRESENTATIONS OR WARRANTIES OF ANY KIND CONCERNING THE WORK, EXPRESS, IMPLIED, STATUTORY OR OTHERWISE, INCLUDING, WITHOUT LIMITATION, WARRANTIES OF TITLE, MERCHANTIBILITY, FITNESS FOR A PARTICULAR PURPOSE, NONINFRINGEMENT, OR THE ABSENCE OF LATENT OR OTHER DEFECTS, ACCURACY, OR THE PRESENCE OF ABSENCE OF ERRORS, WHETHER OR NOT DISCOVERABLE. SOME JURISDICTIONS DO NOT ALLOW THE EXCLUSION OF IMPLIED WARRANTIES, SO SUCH EXCLUSION MAY NOT APPLY TO YOU.
6. Limitation on Liability.
[Bearbeiten]EXCEPT TO THE EXTENT REQUIRED BY APPLICABLE LAW, IN NO EVENT WILL LICENSOR BE LIABLE TO YOU ON ANY LEGAL THEORY FOR ANY SPECIAL, INCIDENTAL, CONSEQUENTIAL, PUNITIVE OR EXEMPLARY DAMAGES ARISING OUT OF THIS LICENSE OR THE USE OF THE WORK, EVEN IF LICENSOR HAS BEEN ADVISED OF THE POSSIBILITY OF SUCH DAMAGES.
7. Termination
[Bearbeiten]- This License and the rights granted hereunder will terminate automatically upon any breach by You of the terms of this License. Individuals or entities who have received Adaptations or Collections from You under this License, however, will not have their licenses terminated provided such individuals or entities remain in full compliance with those licenses. Sections 1, 2, 5, 6, 7, and 8 will survive any termination of this License.
- Subject to the above terms and conditions, the license granted here is perpetual (for the duration of the applicable copyright in the Work). Notwithstanding the above, Licensor reserves the right to release the Work under different license terms or to stop distributing the Work at any time; provided, however that any such election will not serve to withdraw this License (or any other license that has been, or is required to be, granted under the terms of this License), and this License will continue in full force and effect unless terminated as stated above.
8. Miscellaneous
[Bearbeiten]- Each time You Distribute or Publicly Perform the Work or a Collection, the Licensor offers to the recipient a license to the Work on the same terms and conditions as the license granted to You under this License.
- Each time You Distribute or Publicly Perform an Adaptation, Licensor offers to the recipient a license to the original Work on the same terms and conditions as the license granted to You under this License.
- If any provision of this License is invalid or unenforceable under applicable law, it shall not affect the validity or enforceability of the remainder of the terms of this License, and without further action by the parties to this agreement, such provision shall be reformed to the minimum extent necessary to make such provision valid and enforceable.
- No term or provision of this License shall be deemed waived and no breach consented to unless such waiver or consent shall be in writing and signed by the party to be charged with such waiver or consent.
- This License constitutes the entire agreement between the parties with respect to the Work licensed here. There are no understandings, agreements or representations with respect to the Work not specified here. Licensor shall not be bound by any additional provisions that may appear in any communication from You. This License may not be modified without the mutual written agreement of the Licensor and You.
- The rights granted under, and the subject matter referenced, in this License were drafted utilizing the terminology of the Berne Convention for the Protection of Literary and Artistic Works (as amended on September 28, 1979), the Rome Convention of 1961, the WIPO Copyright Treaty of 1996, the WIPO Performances and Phonograms Treaty of 1996 and the Universal Copyright Convention (as revised on July 24, 1971). These rights and subject matter take effect in the relevant jurisdiction in which the License terms are sought to be enforced according to the corresponding provisions of the implementation of those treaty provisions in the applicable national law. If the standard suite of rights granted under applicable copyright law includes additional rights not granted under this License, such additional rights are deemed to be included in the License; this License is not intended to restrict the license of any rights under applicable law.
Creative Commons Notice
[Bearbeiten]Creative Commons is not a party to this License, and makes no warranty whatsoever in connection with the Work. Creative Commons will not be liable to You or any party on any legal theory for any damages whatsoever, including without limitation any general, special, incidental or consequential damages arising in connection to this license. Notwithstanding the foregoing two (2) sentences, if Creative Commons has expressly identified itself as the Licensor hereunder, it shall have all rights and obligations of Licensor.
Except for the limited purpose of indicating to the public that the Work is licensed under the CCPL, Creative Commons does not authorize the use by either party of the trademark "Creative Commons" or any related trademark or logo of Creative Commons without the prior written consent of Creative Commons. Any permitted use will be in compliance with Creative Commons' then-current trademark usage guidelines, as may be published on its website or otherwise made available upon request from time to time. For the avoidance of doubt, this trademark restriction does not form part of the License.
Creative Commons may be contacted at http://creativecommons.org/.
GNU Free Documentation License
[Bearbeiten]Version 1.2, November 2002
Copyright (C) 2000,2001,2002 Free Software Foundation, Inc.
51 Franklin St, Fifth Floor, Boston, MA 02110-1301 USA
Everyone is permitted to copy and distribute verbatim copies
of this license document, but changing it is not allowed.
0. PREAMBLE
The purpose of this License is to make a manual, textbook, or other functional and useful document "free" in the sense of freedom: to assure everyone the effective freedom to copy and redistribute it, with or without modifying it, either commercially or noncommercially. Secondarily, this License preserves for the author and publisher a way to get credit for their work, while not being considered responsible for modifications made by others.
This License is a kind of "copyleft", which means that derivative works of the document must themselves be free in the same sense. It complements the GNU General Public License, which is a copyleft license designed for free software.
We have designed this License in order to use it for manuals for free software, because free software needs free documentation: a free program should come with manuals providing the same freedoms that the software does. But this License is not limited to software manuals; it can be used for any textual work, regardless of subject matter or whether it is published as a printed book. We recommend this License principally for works whose purpose is instruction or reference.
1. APPLICABILITY AND DEFINITIONS
This License applies to any manual or other work, in any medium, that contains a notice placed by the copyright holder saying it can be distributed under the terms of this License. Such a notice grants a world-wide, royalty-free license, unlimited in duration, to use that work under the conditions stated herein. The "Document", below, refers to any such manual or work. Any member of the public is a licensee, and is addressed as "you". You accept the license if you copy, modify or distribute the work in a way requiring permission under copyright law.
A "Modified Version" of the Document means any work containing the Document or a portion of it, either copied verbatim, or with modifications and/or translated into another language.
A "Secondary Section" is a named appendix or a front-matter section of the Document that deals exclusively with the relationship of the publishers or authors of the Document to the Document's overall subject (or to related matters) and contains nothing that could fall directly within that overall subject. (Thus, if the Document is in part a textbook of mathematics, a Secondary Section may not explain any mathematics.) The relationship could be a matter of historical connection with the subject or with related matters, or of legal, commercial, philosophical, ethical or political position regarding them.
The "Invariant Sections" are certain Secondary Sections whose titles are designated, as being those of Invariant Sections, in the notice that says that the Document is released under this License. If a section does not fit the above definition of Secondary then it is not allowed to be designated as Invariant. The Document may contain zero Invariant Sections. If the Document does not identify any Invariant Sections then there are none.
The "Cover Texts" are certain short passages of text that are listed, as Front-Cover Texts or Back-Cover Texts, in the notice that says that the Document is released under this License. A Front-Cover Text may be at most 5 words, and a Back-Cover Text may be at most 25 words.
A "Transparent" copy of the Document means a machine-readable copy, represented in a format whose specification is available to the general public, that is suitable for revising the document straightforwardly with generic text editors or (for images composed of pixels) generic paint programs or (for drawings) some widely available drawing editor, and that is suitable for input to text formatters or for automatic translation to a variety of formats suitable for input to text formatters. A copy made in an otherwise Transparent file format whose markup, or absence of markup, has been arranged to thwart or discourage subsequent modification by readers is not Transparent. An image format is not Transparent if used for any substantial amount of text. A copy that is not "Transparent" is called "Opaque".
Examples of suitable formats for Transparent copies include plain ASCII without markup, Texinfo input format, LaTeX input format, SGML or XML using a publicly available DTD, and standard-conforming simple HTML, PostScript or PDF designed for human modification. Examples of transparent image formats include PNG, XCF and JPG. Opaque formats include proprietary formats that can be read and edited only by proprietary word processors, SGML or XML for which the DTD and/or processing tools are not generally available, and the machine-generated HTML, PostScript or PDF produced by some word processors for output purposes only.
The "Title Page" means, for a printed book, the title page itself, plus such following pages as are needed to hold, legibly, the material this License requires to appear in the title page. For works in formats which do not have any title page as such, "Title Page" means the text near the most prominent appearance of the work's title, preceding the beginning of the body of the text.
A section "Entitled XYZ" means a named subunit of the Document whose title either is precisely XYZ or contains XYZ in parentheses following text that translates XYZ in another language. (Here XYZ stands for a specific section name mentioned below, such as "Acknowledgements", "Dedications", "Endorsements", or "History".) To "Preserve the Title" of such a section when you modify the Document means that it remains a section "Entitled XYZ" according to this definition.
The Document may include Warranty Disclaimers next to the notice which states that this License applies to the Document. These Warranty Disclaimers are considered to be included by reference in this License, but only as regards disclaiming warranties: any other implication that these Warranty Disclaimers may have is void and has no effect on the meaning of this License.
2. VERBATIM COPYING
You may copy and distribute the Document in any medium, either commercially or noncommercially, provided that this License, the copyright notices, and the license notice saying this License applies to the Document are reproduced in all copies, and that you add no other conditions whatsoever to those of this License. You may not use technical measures to obstruct or control the reading or further copying of the copies you make or distribute. However, you may accept compensation in exchange for copies. If you distribute a large enough number of copies you must also follow the conditions in section 3.
You may also lend copies, under the same conditions stated above, and you may publicly display copies.
3. COPYING IN QUANTITY
If you publish printed copies (or copies in media that commonly have printed covers) of the Document, numbering more than 100, and the Document's license notice requires Cover Texts, you must enclose the copies in covers that carry, clearly and legibly, all these Cover Texts: Front-Cover Texts on the front cover, and Back-Cover Texts on the back cover. Both covers must also clearly and legibly identify you as the publisher of these copies. The front cover must present the full title with all words of the title equally prominent and visible. You may add other material on the covers in addition. Copying with changes limited to the covers, as long as they preserve the title of the Document and satisfy these conditions, can be treated as verbatim copying in other respects.
If the required texts for either cover are too voluminous to fit legibly, you should put the first ones listed (as many as fit reasonably) on the actual cover, and continue the rest onto adjacent pages.
If you publish or distribute Opaque copies of the Document numbering more than 100, you must either include a machine-readable Transparent copy along with each Opaque copy, or state in or with each Opaque copy a computer-network location from which the general network-using public has access to download using public-standard network protocols a complete Transparent copy of the Document, free of added material. If you use the latter option, you must take reasonably prudent steps, when you begin distribution of Opaque copies in quantity, to ensure that this Transparent copy will remain thus accessible at the stated location until at least one year after the last time you distribute an Opaque copy (directly or through your agents or retailers) of that edition to the public.
It is requested, but not required, that you contact the authors of the Document well before redistributing any large number of copies, to give them a chance to provide you with an updated version of the Document.
4. MODIFICATIONS
You may copy and distribute a Modified Version of the Document under the conditions of sections 2 and 3 above, provided that you release the Modified Version under precisely this License, with the Modified Version filling the role of the Document, thus licensing distribution and modification of the Modified Version to whoever possesses a copy of it. In addition, you must do these things in the Modified Version:
- A. Use in the Title Page (and on the covers, if any) a title distinct from that of the Document, and from those of previous versions (which should, if there were any, be listed in the History section of the Document). You may use the same title as a previous version if the original publisher of that version gives permission.
- B. List on the Title Page, as authors, one or more persons or entities responsible for authorship of the modifications in the Modified Version, together with at least five of the principal authors of the Document (all of its principal authors, if it has fewer than five), unless they release you from this requirement.
- C. State on the Title page the name of the publisher of the Modified Version, as the publisher.
- D. Preserve all the copyright notices of the Document.
- E. Add an appropriate copyright notice for your modifications adjacent to the other copyright notices.
- F. Include, immediately after the copyright notices, a license notice giving the public permission to use the Modified Version under the terms of this License, in the form shown in the Addendum below.
- G. Preserve in that license notice the full lists of Invariant Sections and required Cover Texts given in the Document's license notice.
- H. Include an unaltered copy of this License.
- I. Preserve the section Entitled "History", Preserve its Title, and add to it an item stating at least the title, year, new authors, and publisher of the Modified Version as given on the Title Page. If there is no section Entitled "History" in the Document, create one stating the title, year, authors, and publisher of the Document as given on its Title Page, then add an item describing the Modified Version as stated in the previous sentence.
- J. Preserve the network location, if any, given in the Document for public access to a Transparent copy of the Document, and likewise the network locations given in the Document for previous versions it was based on. These may be placed in the "History" section. You may omit a network location for a work that was published at least four years before the Document itself, or if the original publisher of the version it refers to gives permission.
- K. For any section Entitled "Acknowledgements" or "Dedications", Preserve the Title of the section, and preserve in the section all the substance and tone of each of the contributor acknowledgements and/or dedications given therein.
- L. Preserve all the Invariant Sections of the Document, unaltered in their text and in their titles. Section numbers or the equivalent are not considered part of the section titles.
- M. Delete any section Entitled "Endorsements". Such a section may not be included in the Modified Version.
- N. Do not retitle any existing section to be Entitled "Endorsements" or to conflict in title with any Invariant Section.
- O. Preserve any Warranty Disclaimers.
If the Modified Version includes new front-matter sections or appendices that qualify as Secondary Sections and contain no material copied from the Document, you may at your option designate some or all of these sections as invariant. To do this, add their titles to the list of Invariant Sections in the Modified Version's license notice. These titles must be distinct from any other section titles.
You may add a section Entitled "Endorsements", provided it contains nothing but endorsements of your Modified Version by various parties--for example, statements of peer review or that the text has been approved by an organization as the authoritative definition of a standard.
You may add a passage of up to five words as a Front-Cover Text, and a passage of up to 25 words as a Back-Cover Text, to the end of the list of Cover Texts in the Modified Version. Only one passage of Front-Cover Text and one of Back-Cover Text may be added by (or through arrangements made by) any one entity. If the Document already includes a cover text for the same cover, previously added by you or by arrangement made by the same entity you are acting on behalf of, you may not add another; but you may replace the old one, on explicit permission from the previous publisher that added the old one.
The author(s) and publisher(s) of the Document do not by this License give permission to use their names for publicity for or to assert or imply endorsement of any Modified Version.
5. COMBINING DOCUMENTS
You may combine the Document with other documents released under this License, under the terms defined in section 4 above for modified versions, provided that you include in the combination all of the Invariant Sections of all of the original documents, unmodified, and list them all as Invariant Sections of your combined work in its license notice, and that you preserve all their Warranty Disclaimers.
The combined work need only contain one copy of this License, and multiple identical Invariant Sections may be replaced with a single copy. If there are multiple Invariant Sections with the same name but different contents, make the title of each such section unique by adding at the end of it, in parentheses, the name of the original author or publisher of that section if known, or else a unique number. Make the same adjustment to the section titles in the list of Invariant Sections in the license notice of the combined work.
In the combination, you must combine any sections Entitled "History" in the various original documents, forming one section Entitled "History"; likewise combine any sections Entitled "Acknowledgements", and any sections Entitled "Dedications". You must delete all sections Entitled "Endorsements."
6. COLLECTIONS OF DOCUMENTS
You may make a collection consisting of the Document and other documents released under this License, and replace the individual copies of this License in the various documents with a single copy that is included in the collection, provided that you follow the rules of this License for verbatim copying of each of the documents in all other respects.
You may extract a single document from such a collection, and distribute it individually under this License, provided you insert a copy of this License into the extracted document, and follow this License in all other respects regarding verbatim copying of that document.
7. AGGREGATION WITH INDEPENDENT WORKS
A compilation of the Document or its derivatives with other separate and independent documents or works, in or on a volume of a storage or distribution medium, is called an "aggregate" if the copyright resulting from the compilation is not used to limit the legal rights of the compilation's users beyond what the individual works permit. When the Document is included in an aggregate, this License does not apply to the other works in the aggregate which are not themselves derivative works of the Document.
If the Cover Text requirement of section 3 is applicable to these copies of the Document, then if the Document is less than one half of the entire aggregate, the Document's Cover Texts may be placed on covers that bracket the Document within the aggregate, or the electronic equivalent of covers if the Document is in electronic form. Otherwise they must appear on printed covers that bracket the whole aggregate.
8. TRANSLATION
Translation is considered a kind of modification, so you may distribute translations of the Document under the terms of section 4. Replacing Invariant Sections with translations requires special permission from their copyright holders, but you may include translations of some or all Invariant Sections in addition to the original versions of these Invariant Sections. You may include a translation of this License, and all the license notices in the Document, and any Warranty Disclaimers, provided that you also include the original English version of this License and the original versions of those notices and disclaimers. In case of a disagreement between the translation and the original version of this License or a notice or disclaimer, the original version will prevail.
If a section in the Document is Entitled "Acknowledgements", "Dedications", or "History", the requirement (section 4) to Preserve its Title (section 1) will typically require changing the actual title.
9. TERMINATION
You may not copy, modify, sublicense, or distribute the Document except as expressly provided for under this License. Any other attempt to copy, modify, sublicense or distribute the Document is void, and will automatically terminate your rights under this License. However, parties who have received copies, or rights, from you under this License will not have their licenses terminated so long as such parties remain in full compliance.
10. FUTURE REVISIONS OF THIS LICENSE
The Free Software Foundation may publish new, revised versions of the GNU Free Documentation License from time to time. Such new versions will be similar in spirit to the present version, but may differ in detail to address new problems or concerns. See http://www.gnu.org/copyleft/.
Each version of the License is given a distinguishing version number. If the Document specifies that a particular numbered version of this License "or any later version" applies to it, you have the option of following the terms and conditions either of that specified version or of any later version that has been published (not as a draft) by the Free Software Foundation. If the Document does not specify a version number of this License, you may choose any version ever published (not as a draft) by the Free Software Foundation.